政策研のページ ドラッグ・ラグ:なぜ、未承認薬が増えているのか?
2010年代後半に国内未承認薬が増加していることが政策研ニュース※1や国立研究開発法人国立がん研究センターの調査※2にて明らかになっています。「なぜ、未承認薬が増えているのか」の問いに答えるため、増加の要因を抗悪性腫瘍剤および神経系用剤について分析したところ、新興企業の品目が顕著に増加したこと、ピボタル試験の臨床試験相が早期化したこと(抗悪性腫瘍剤)、その品目の米国承認時のピボタル試験に日本地域の組入れが少ないことが明らかになりました。本稿では、増加要因を分析した政策研ニュースNo.66「ドラッグ・ラグ:なぜ、未承認薬が増えているのか?」(2022年7月)の概要を紹介します。
1. はじめに
2010年代後半に未承認薬数が増加していました。その実態として日本法人や国内管理人をもたない新興バイオ医薬品企業の製品が多く、国内では開発に着手されていないことが示唆されています※1※3。また、これら未承認薬には臨床的に重要度の高い医薬品が多くを占めることがわかっています※4。未承認薬の増加は、日本の最新医薬品アクセスにとって見過ごせない課題として、その原因と対策の検討が必要とされます。
2011年から2020年の抗悪性腫瘍剤および神経系用剤の米国食品医薬品局(FDA)承認薬(新規有効成分)を対象に日本未承認薬の特徴を分析し、未承認薬増加の背景とともにその要因を追究することで、未承認薬増加の解消に向けた方向性を考察します。
-
※1医薬産業政策研究所「ドラッグ・ラグ:国内未承認薬の状況とその特徴」政策研ニュースNo.63(2021年7月)
-
※2内閣官房 健康・医療戦略推進本部「第1回 医薬品開発協議会 資料2-6」(2020年10月27日)
-
※3IQVIA “Global Trends in R&D: Overview through 2020”(May 2021)
-
※4医薬産業政策研究所「ドラッグ・ラグ:未承認薬は日本のアンメット・メディカル・ニーズに応えうるか?」政策研ニュースNo.66(2022年7月)
2. 研究方法
FDAにて2011年から2020年に承認された品目のうち、未承認薬が多い疾患領域※1※4の抗悪性腫瘍剤(104品目)および神経系用剤(32品目)を分析対象としました。2011年から2015年を前期、2016年から2020年を後期として分けて比較し、前期は2015年末時点、後期は2020年末時点とし、日本では承認されていない品目を未承認薬としました。FDA承認時のピボタル試験※5はPrescribing Informationから同定し、その臨床試験情報はClinicalTrials.govから取得しました※6※7。企業の分類はEvaluate Pharmaにて調査し、新興企業は1990年以降に設立した企業とし、それ以外を製薬企業としました。承認薬増加への影響度は、統計解析手法を用いて、ロジスティック回帰分析および最小二乗法を用いた線形モデルにより推定しました。
-
※5医薬産業政策研究所「抗悪性腫瘍剤と神経系用剤におけるFDA承認動向の変化 —日本の未承認薬増加の背景—」政策研ニュースNo.66(2022年7月)
-
※6医薬産業政策研究所「近年の国際共同治験の参加国の分析 —臨床試験登録システムClinicalTrials.govを基に—」政策研ニュースNo.58(2019年11月)
-
※7医薬産業政策研究所「近年における国際共同治験の動向調査」政策研ニュースNo.66(2022年7月)
3. 分析結果
3-1. 日本未承認薬の内訳:抗悪性腫瘍剤
調査期間中に、FDAで承認された抗悪性腫瘍剤は、それぞれ前期44品目、後期60品目ありました。そのうち、日本未承認薬はそれぞれ前期27品および後期41品あり、日本未承認薬比率は61%から68%に増加していました(図1)。抗悪性腫瘍剤においても、先の報告※1と同様に増加したことが確認されました。
図1 抗悪性腫瘍剤の未承認薬数・比率の変化
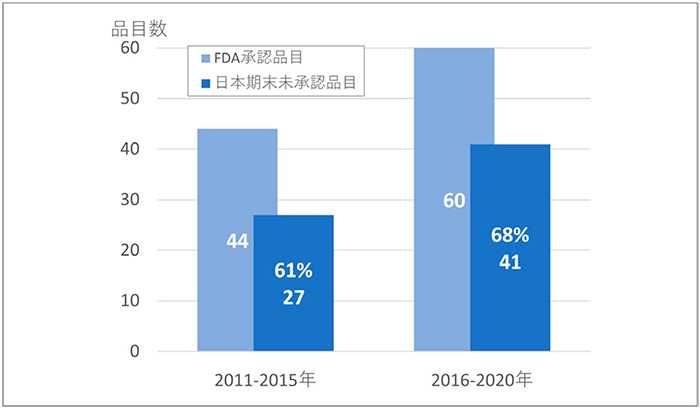
出所:FDA、独立行政法人医薬品医療機器総合機構(PMDA)の公開情報をもとに医薬産業政策研究所にて作成
図2では、FDAへの申請者を新興企業もしくは製薬企業に分類して未承認薬数の変化を示しています。新興企業の品目では、承認薬は2品目から3品目に微増でしたが、日本未承認薬は6品目から22品目に顕著に増加していました。製薬企業の品目では、承認薬および未承認薬ともに前期と後期に品目数の大きな変化は見られておらず、日本未承認薬の増加は、新興企業品目の増加であることがわかりました。
図2 企業分類別未承認薬数
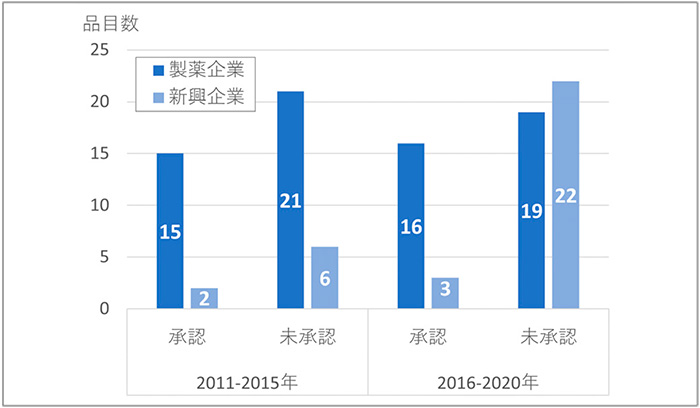
出所:FDA、PMDAの公開情報、Evaluate Pharmaをもとに医薬産業政策研究所にて作成
海外に遅れることなく日本で承認されるためには、海外承認時のピボタル試験が国際共同治験であり、その試験に日本地域が組み入れられていることが重要となります。図3では、FDA承認時のピボタル試験に日本地域が組み入れられているかを調べた結果を示しています。まず、国際共同治験について見ますと、前期は43品目(98%)、後期では53品目(88%)となり、後期では米国での単国試験が増加し、国際共同治験比率が低下していました。日本地域が組み入れられているピボタル試験は、前期では11品目(25%)、後期では21品目(35%)と品目数・組入れ率ともに増加していました。日本への最新薬剤のアクセスを確かなものにするため、「国際共同治験に関する基本的考え方」(2007年、厚生労働省)の発出、国際共同治験への日本地域組入れにおける各製薬企業の尽力、審査期間短縮への審査当局の功績があり※6※7、一定の成果が見られていました。しかし、日本組入れがないピボタル試験の比率(65%、39品目)は依然として高く、承認ラグや未承認薬となる可能性が危惧されました。
図3 抗悪性腫瘍剤のピボタル試験への日本地域組入れ状況
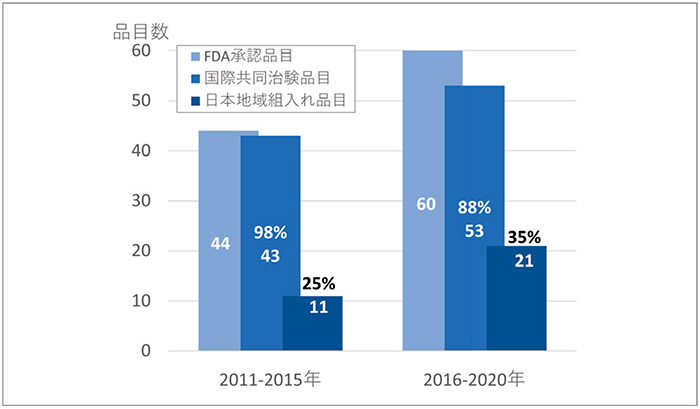
注:2ヵ国以上を組み入れている試験を国際共同臨床試験とした。複数の試験がある場合は後期臨床相を集計した
出所:FDA、PMDAの公開情報、ClinicalTrials.govをもとに医薬産業政策研究所にて作成
図4では、日本地域組入れの有無に分類し、未承認薬数の変化を示しています。
日本未承認薬では、日本地域組入れ有りは4品目から6品目に増加しましたが、組入れ無しは23品目から35品目に顕著に増加しました。後期の未承認薬においては、組入れ無しの比率は、85%(35/41品目)に達していました。なお、承認薬では、日本組入れ有りは7品目から15品目に倍増し、組入れ無しは10品目から4品目に減少し、国際共同治験への日本組入れが進んでいました。
日本未承認薬の増加は、日本地域の組入れが無いピボタル試験の増加であることがわかりました。
図4 ピボタル試験の日本地域組入れ状況
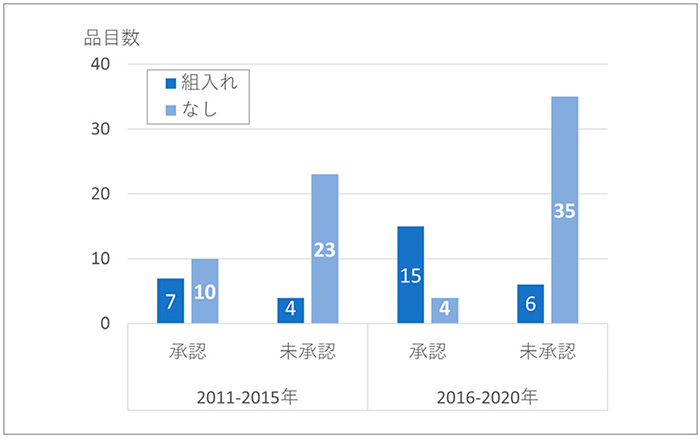
注:2ヵ国以上を組み入れている試験を国際共同臨床試験とした。
出所:FDA、PMDAの公開情報、ClinicalTrials.govをもとに医薬産業政策研究所にて作成
ピボタル試験にどんな国や地域が組み入れられているか、後期の60品目について調べました(図5)。日本承認薬19品目では18品目が国際共同治験であり、日本の組入れ品目数(15品目)はG7各国と同等でした。韓国(13品目)や台湾(10品目)も多く組み入れられており、アジア諸国においても国際共同治験が推進されていました。
日本未承認41品目では、34品目は国際共同治験、6品目は単一国試験でした。日本を除くG7各国では30品目程度に組み入れられているのに対し、日本は6品目にとどまっていました。アジアでは、韓国(16品目)および台湾(11品目)が組み入れられており、日本地域の組入れは韓国や台湾より少ないことが示されました。人口が少なく、医薬品市場も小さい韓国・台湾より組入れ数が少ないことは、日本地域の薬事・臨床試験環境に相違があることが示唆されました。新興企業の未承認品目では、21品目のうち19品目は国際共同治験であり、組入れ国の傾向は未承認薬全体と同様でした。
図5 2016~2020年の抗悪性腫瘍剤 ピボタル試験への組入れ地域
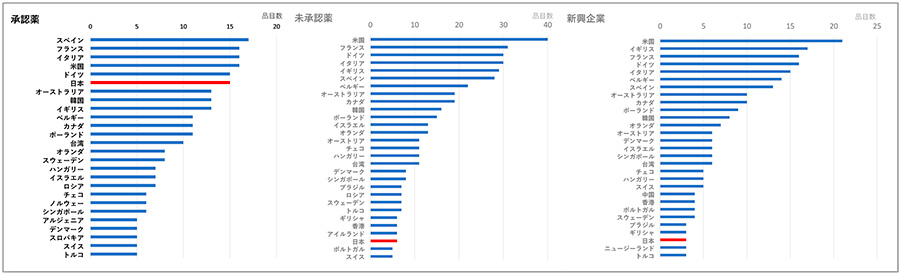
注:2016~2020年のFDA承認薬のうち、2020年末時点の日本承認薬(19品目、左図)と日本未承認薬(41品目、中図)のピボタル試験への組入れが5品目以上の地域を示す。未承認薬のうち、新興企業の品目(21品目、右図)は3品目以上の地域を示す。
出所:FDA、PMDAの各公開情報、Evaluate Pharma、ClinicalTrials.govをもとに医薬産業政策研究所にて作成
FDA承認品目の後期では、早期臨床試験相(Phase 1からPhase 2)のピボタル試験が増えています[5]。前期から後期にかけての臨床試験相の変化を見ますと(図6)、未承認薬では、Phase 3は13品目で横ばいのところ、Phase 2(13から18品目)やPhase 1/2(1から8品目)は増加していました。後期ではPhase 1の2品目も見られました。日本承認薬においてもPhase 2やPhase 1/2がそれぞれ6品目および2品目に増加しましたが、Phase 3の品目数(11品目)は減少しました。
未承認薬増加は、Phase 3の品目数は13品目から変化がなかったことから、早期臨床試験相(Phase 2、Phase 1/2、Phase 1)の品目数の増加(14品目から28品目)であることがわかりました。
図6 臨床試験相別未承認薬数
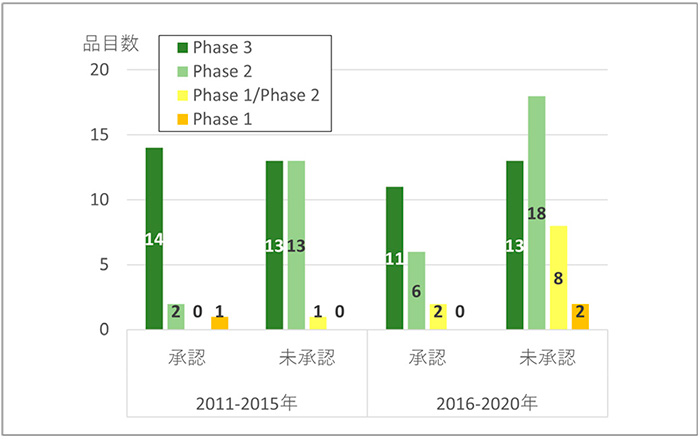
注:ピボタル試験が複数ある場合、後期相の試験を集計
出所:FDA、PMDAの公開情報、Evaluate pharma、ClinicalTrials.govをもとに医薬産業政策研究所にて作成
未承認薬の増加の内訳として、1)新興企業品目の増加、2)ピボタル試験相の早期化、3)ピボタル試験相への低い日本組入れ率が起こっていることがわかりました。
これらの要素について、未承認薬の増加への影響の大きさを統計手法により推定しました(表1)。
表1 未承認薬増加への影響度
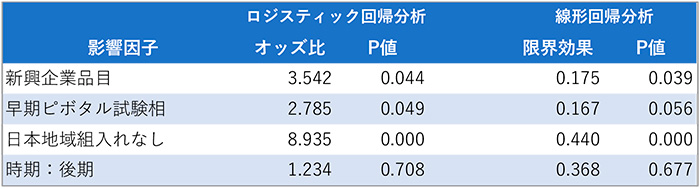
出所:医薬産業政策研究所にて作成
104品目のデータを用いたロジスティック回帰分析では、「新興企業品目」「早期ピボタル試験相」「日本地域組入れ無し」、と3つの影響因子において統計学的な有意差が認められました。日本地域を組み入れない場合は組み入れる場合に比べて、未承認薬となるオッズ比が8.9倍と推定され、影響が大きいことが示されました。また、最小二乗法を用いた線形確率モデルによる推定においても、いずれも統計学的な有意性が認められました。日本地域を組み入れない場合は組み入れる場合に比べて、未承認薬になる確率が44%高いことがわかり、新興企業品目や早期のピボタル試験相ではそれぞれ18%および17%であったことから、日本地域の組入れ無しは、評価した3つの因子の中では未承認薬となる確率が最も高いことが示されました。以上の統計解析結果から、未承認薬増加の要因として、ピボタル試験へ日本組入れがないことの影響が大きく、次いで新興企業、早期臨床試験相であることがわかりました。
未承認増加の内訳を企業分類とピボタル臨床試験相にそれぞれ分解し、詳細に分析しました(図7)。新興企業で前期(6品目)から後期(22品目)への16品目の増加内訳は、Phase 2が11品目に、Phase 1/2およびPhase 1で6品目になり、早期臨床試験相で計17品目(77%)と前期から13品目の増加が見られました。製薬企業の未承認薬では、品目数は大きな増減はありませんでしたが、早期臨床相の比率が58%(計11品目)と前期に比べて増加していました。
図7 企業分類別ピボタル試験相
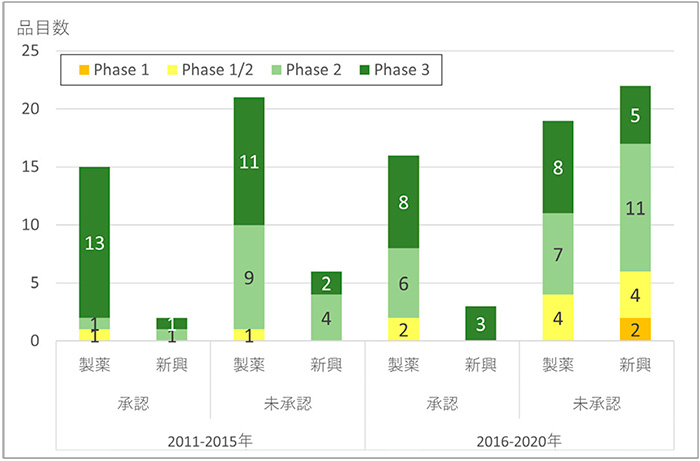
注:ピボタル試験が複数ある場合、後期相の試験を集計
出所:FDA、PMDAの公開情報、Evaluate Pharma、ClinicalTrials.govをもとに医薬産業政策研究所にて作成
さらに企業分類とピボタル臨床試験相における日本地域の組入れ状況を分析しました(図8)。
製薬企業の組入れは、前期計10品目(28%)から後期計15品目(43%)と、品目数・組入れ率ともに増加していました。新興企業においても前期1品目(13%)から後期計6品目(24%)に増加していたものの、製薬企業の品目に比べて組入れ率は低いものでした。臨床試験相別に組入れ率を見ると、製薬企業のPhase 3では7品目(29%)から9品目(56%)と、後期には品目数・率ともに増加していました。Phase 2の組入れ率においても2品目(20%)から5品目(38%)へ増加していました。その一方、後期におけるPhase 1/2への組入れ率は17%(1/6品目)と低い値でした。新興企業では、前期では1品目であったのに対し、後期では、Phase 3への組入れは3品目(38%)、Phase 2では2品目(18%)、Phase 1/2では1品目(25%)、Phase 1では0品目(0%、0/2品目)と早期相への組入れが少ない結果でした。
前期から後期にかけて、ピボタル試験への日本組入れ率は増加していたものの、新興企業の品目および早期臨床試験相で日本地域の組入れ率が後期で低いことが明らかとなりました。
図8 臨床試験相別、企業分類別の日本組入れ
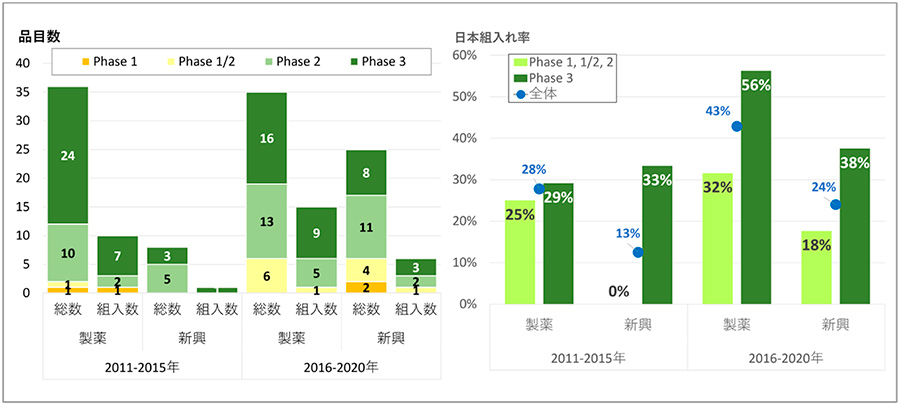
注:ピボタル試験が複数ある場合、後期相の試験を集計、%は組入れ率を示す
出所:FDA、PMDAの各公開情報、Evaluate Pharma、ClinicalTrials.govをもとに医薬産業政策研究所にて作成
3-2. 日本未承認薬の内訳:神経系用剤
2011~2015年および2016~2020年にFDAで承認された神経系用剤についても日本未承認薬の状況を調査しました(図9)。神経系用剤のFDA承認品目数は12品目から30品目へと2倍以上に増加していました。日本未承認薬は、FDA承認薬数の増加に比例し、9品目(75%)から22品目(73%)に大きく増加し、未承認薬の品目数の増加が確認されました。ピボタル試験への日本組入れ状況は、前期で3品目(25%)、後期で6品目(20%)と組入れ品目は増えましたが、組入れ比率は低下し、また、いずれの時期でも低い組入れ率でした。
図9 神経系用剤の未承認薬
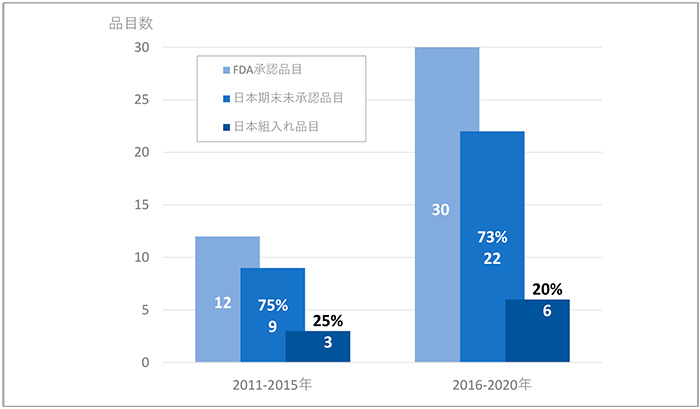
注:ピボタル試験が複数ある場合、後期相の試験を集計
出所:FDA、PMDAの公開情報、Evaluate Pharma、ClinicalTrials.govをもとに医薬産業政策研究所にて作成
神経系用剤におけるFDA承認状況やピボタル試験への日本組入れを企業分類ごとに分解してみます(図10)。FDA承認品目では、前期は製薬企業9品目(75%)、新興企業3品目と製薬企業の品目が主体でしたが、後期は製薬企業10品目(33%)、新興企業20品目(67%)と主体は新興企業の品目に移っていました。日本未承認薬では、製薬企業で品目数・未承認薬比率に変化はありませんでしたが、新興企業で2品目(67%)から14品目(70%)と未承認薬の品目数が大幅に増加していました。ピボタル試験への日本地域の組入れは、製薬企業で3品目(33%)から4品目(40%)に、新興企業で0品目から2品目(10%)に微増していましたが、日本組入れ比率は製薬企業で40%、新興企業では10%と新興企業での組入れ率が特に低いことがわかりました。
神経系用剤のFDA承認薬の未承認薬数の増加は、抗悪性腫瘍剤と同様に、後期において新興企業の品目が増加し、日本地域が組入れられていないことが未承認薬増加の要因であることがわかりました。
図10 企業別の神経系用剤の未承認薬
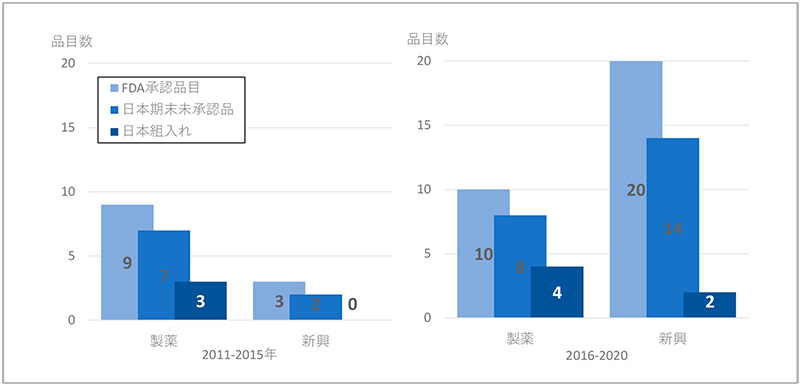
注:ピボタル試験が複数ある場合、後期相の試験を集計
出所:FDA、PMDAの公開情報、Evaluate Pharma、ClinicalTrials.govをもとに医薬産業政策研究所にて作成
3-3. 新興企業の事業における特徴と環境
新興企業の実態に迫るため、2011~2020年に抗悪性腫瘍剤をFDAに申請した新興企業29社(33品目)のうち、企業情報取得が可能な26社について、会社設立からFDA承認までの事業経緯を追いました。
26社の内訳は、23社(88%)が米国企業、他の3社はアイルランド、ドイツ、中国の企業であり、その多くは米国拠点の企業であることが確認されました。26社個社ごとに会社設立からFDA承認までの期間を求め、中央値を算出すると、設立から上場まで4年3ヵ月となり、設立からFDA承認までは14年11ヵ月、上場後からFDA承認まででも6年9ヵ月の時間を要していることが明らかになりました。また、FDA承認の前年と5年前での新興企業の医薬品売上を見ますと、FDA承認前年で17社(65%)、FDA承認5年前で21社(81%)において医薬品売上はありませんでした(図11)。
新興企業では、会社設立から新薬の上市までは約15年の時間を要するとともに、承認前ではほとんどの企業で売上収益がないことが確認されました。これらの企業では長期間にわたり研究開発を進めるための資金は潤沢ではないことが改めて認識されました。
図11 抗悪性腫瘍剤の新興企業の売上状況
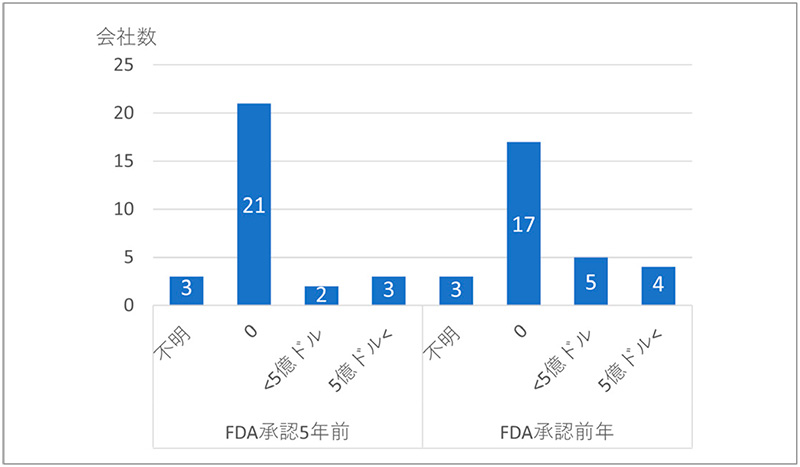
注:2011~2020年にFDA承認の抗悪性腫瘍剤を有する新興企業29社のデータを示す。
出所:FDAの公開情報、Evaluate Pharmaをもとに医薬産業政策研究所にて作成
9割方は米国企業である新興企業にとっては、米国での将来的な収益最大化は第一優先であることには違いありませんが、米国以外の各国をピボタル試験に組み入れることについては、研究開発の効率性とともに製品として成功した際の各国での期待収益のバランス、いわゆる投資対効果を視野に入れていることが考えられます。そこで各国の医薬品市場は、米国新興企業からどのように映っているかを類推するため、米国市場に対する相対的な市場規模の推移を描出しました(表2)。米国の医薬品市場規模を該当年ごとに100とした際に、各国あるいは各地域の医薬品市場規模を指数化すると、中国のみで成長性(年平均成長率4.7%)が見られ、欧州(-3.1%)と日本(-6.1%)はマイナス成長でした。規模では、欧州は2020年でも米国の半分以上(56)はありましたが、日本(16)に至っては米国の7分の1強程度の小さい規模でした。日本の市場は規模も小さく、マイナス成長に映ることが示され、日本の薬剤市場は魅力的なマーケットとは捉えがたい市場であり、新興企業にとって投資優先度は低い可能性が推察されました。
表2 米国の医薬品市場に対する、相対的市場規模推移
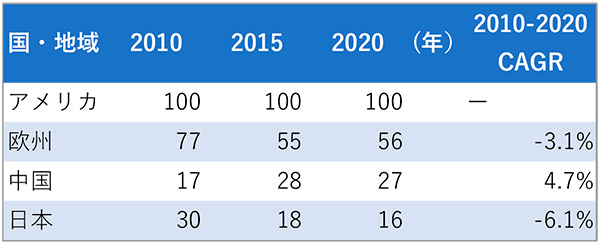
注:市場規模は、米国販売額を100とした場合の各国・地域の販売額を指数化した。
出所:Copyright(c)2022 IQVIA. IQVIA World Review, Data Period - Year 2010- 2020をもとに医薬産業政策研究所にて作成(無断転載禁止)
ほとんどの新興企業は日本国内に開発母体をもっておりません。日本国内への展開状況を確認するため、新興企業品目の日本国内開発状況(2022年4月末時点)を調査しました。2011~2020年にFDAで承認された抗悪性腫瘍剤および神経系用剤のうち、新興企業品目はそれぞれ33品目(29社)および23品目(21社)ありました。新興企業品目は、FDA承認取得を境に、大きく3通りのビジネス形態がとられ、1つ目は製薬企業によるM&A、2つ目は日本テリトリーの製品導出、3つ目は自社による日本市場開拓等でした(図12)。
抗悪性腫瘍剤の33品目(29社)についてビジネス形態ごとに開発状況を見ますと、M&Aの11品目(9社)については、日本企業1社を含む、いわゆるグローバル製薬企業によりFDA承認の1~2年後に買収され、国内開発状況は承認(8品目)あるいは開発中(3品目)でした。導出の9品目はグローバルあるいは日本テリトリーにおいてFDA承認の1~2年後に主に日本の製薬企業に導出され、承認(4品目)あるいは開発中(5品目)でした。M&Aの形態に比べると承認に至っている割合は低いようでした。自社の13品目では、複数製品をもつ新興企業では日本法人を設立し、承認2品目あるいは開発中2品目と国内開発が進められていました。しかし多くの品目は、情報無し(9品目)と、2022年4月末時点で開発されている様子は確認できませんでした。なお、情報無しは調査したデータベースに記載がないことに加え、国内臨床試験を実施した経緯はあるが続報がない、あるいは中止の品目を含むため、医薬品の科学的な有用性が検証されなかった可能性も推察されました。
新興企業の品目では、M&Aや導出により日本での商業化が進む一方、企業-企業間の交渉等のプロセス期間もまた承認ラグとなり得ることが示唆されました。また、情報無しが9品目残っており、ドラッグロスとなるリスクが見られました。
図12 新興企業の抗悪性腫瘍剤品目の国内状況
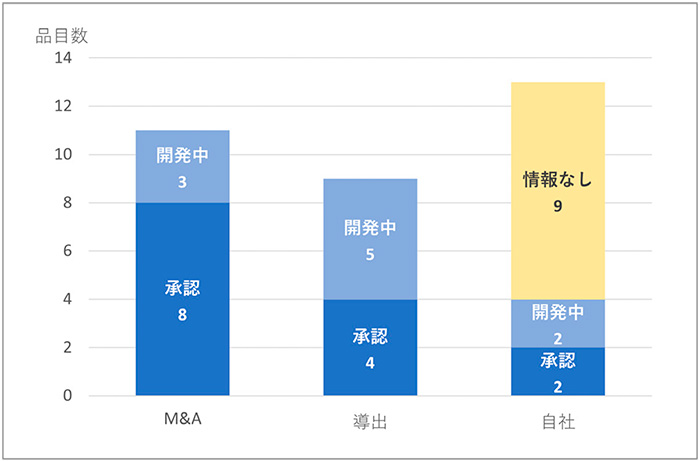
注:2011~2020年にFDA承認の抗悪性腫瘍剤33品目(新興企業29社)について、2021年末時点の開発状況を示す。
出所:FDAの公開情報、Evaluate Pharma、明日の新薬をもとに医薬産業政策研究所にて作成
神経系用剤23品目(21社)では(図13)、M&Aは1品目、導出は11品目で見られ、いずれも承認あるいは開発されていました。自社の分類では、11品目中1品目のみで国内開発されていましたが、10品目では国内で臨床開発が実施されている情報は確認されず、ドラッグロスとなるリスクが見られました。
図13 新興企業の神経系用剤品目の国内状況
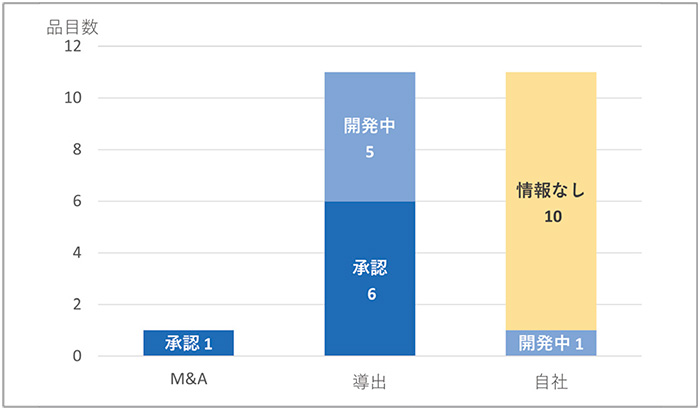
注:2011~2020年にFDA承認の神経系用剤23品目(新興企業21社)について、2021年末時点の開発状況を示す。
出所:FDAの公開情報、Evaluate Pharma、明日の新薬をもとに医薬産業政策研究所にて作成
4. 考察
2016~2020年における未承認薬増加の最も影響の大きい因子として、ピボタル試験への日本組入れがないことが挙げられました。特に新興企業のFDA承認品目増加に伴い、新興企業品目のピボタル試験への組入れ率が低いことは大きな要因でした。「新興企業のピボタル試験にはなぜ、日本は組み入れられないのであろうか?」の疑問に対して、新興企業の投資意思決定の側面:研究開発と期待事業価値の側面から考察します。
抗悪性腫瘍剤のほとんどの品目では国際共同治験を実施していましたが、日本地域の組入れ優先度が低いようでした。人口は少なく、医薬品市場も小さい韓国、台湾、香港、シンガポール等のアジア諸国が多く組み入れられていたことから、患者登録数・登録速度、試験開始にあたっての手続きの煩雑さ(日本語書類の整備を含め)、臨床試験の費用等に臨床試験環境の面で日本地域を選びにくい可能性が考えられました。また、国際共同治験に日本が組み入れられる際には、薬事規制上、日本人での忍容性評価等の追加的な臨床試験を事前に行う必要があり、組入れのハードルになっている可能性があります。新興企業から見た日本地域における薬事・臨床試験にかかわる課題については、より深い精査が必要と思われます。
新興企業が日本市場に早く事業展開する場合や、M&Aおよび導出時に事業価値を高めたい場合には、国際共同治験に日本を組み入れるモチベーションがあることが想像されます。しかしながら、ここ10年の日本の相対的な市場規模は縮小し、マイナス成長の市場と映っていることが類推され、投資資源が限られた新興企業においては、日本事業展開への投資優先度は低いことが考えられました。新興企業がピボタル試験に日本地域を組み入れていない理由については、公開情報からの推察には限界があり、個社ごとの実態聞き取り調査等が必要と思われます。
新興企業の品目の日本事業展開の実態は、FDA承認の1~2年後にM&Aや導出により、日本での事業展開が図られていました。製薬企業や日本企業による新興企業との早期の事業提携、あるいは製品導入時の判断には、経済合理性の観点から、日本の医薬品市場の魅力度が重要であることが考えられます。
2010年以前に採り上げられたドラッグ・ラグの課題は、国際共同治験への日本地域参加が製薬企業により推進され、また、薬事承認の審査期間についても短縮が図られ、大きく改善してきました。今回、新興企業の品目が増えたことにより、新たなドラッグ・ラグの兆候が示され、その課題はより複雑化してきました。未承認薬増加の主要因である「新興企業のピボタル試験に日本地域が組み入れられない」ことの理由までは明らかにはなっていませんが、これを解消するためには、「新興企業品目の国際共同治験に日本地域が組み入れられるためにはどうするべきか」あるいは、「新興企業と日本・欧米の製薬企業が、早くから日本市場展開で提携するためにはなにが必要か」が命題であることが考えられます。これらの課題に対しては、企業努力を超えて、海外の新興企業が日本を組み入れるインセンティブを提供する政策や、製薬企業が日本で事業展開する際に収益を担保できるための政策が重要と考えます。日本への最新医薬品のアクセスを確保するためには、世界の医薬品開発状況の変化を鑑みた政策の議論が進むことを期待します。
さらに、日本への最新医薬品アクセスを最重要視する際には、日本人データを用いずに、海外での有用性検証のデータでの承認を許容する等の規制の見直しも必要となるかもしれません。個別化医療が進み、個々人に合わせた薬剤が増える中、民族的要因や環境要因に過剰に留意することなく、有用性が評価できるような科学の進展とともに、薬事規制のあり方についても議論が進むことを切に願います。
(医薬産業政策研究所 統括研究員 飯田 真一郎)