Topics
ドラッグ・ラグ:小児適応を持つ日本未承認薬の特徴
その2 -抗がん剤-
医薬産業政策研究所 主任研究員 東 宏
医薬産業政策研究所 統括研究員 飯田真一郎
1. はじめに
これまで医薬産業政策研究所ではドラッグ・ラグの現状分析を重ねてきた1、2、3、4、5)。ドラッグ・ラグとは、海外での既承認薬が日本国内での薬事承認を得るまでに年月を要するという問題のことであり、2つの側面を有する。日本で承認されているが他国よりも時間を要した「ラグ(遅延)」という側面と、他国で承認されているが日本では承認されていない「日本未承認」という側面である。この日本未承認は長期間放置されると、「ドラッグ・ロス」となり、重大な問題となり得る。
日本未承認薬が関連する小児疾患の多くは希少疾患であり、また未承認の疾病領域にがんが多いことが報告されている3)。厚生労働省でも小児がんの医薬品開発推進に力を入れ、調査を進めている6)。本稿では、小児がん用医薬品を抗がん剤のうち小児適応を取得しているものと定義し、日米の承認および適応状況を調査のうえ、ドラッグ・ラグの状況について報告する。またドラッグ・ラグの要因解明に繋げるべく、米国における開発担い手やピボタル試験(薬事承認申請に必要なデータを取得する検証的試験)を分析し報告する。
2. 調査方法
本稿では、米国での医薬品の承認及び小児適応取得状況として、U. S. Food and Drug Administration(FDA)がHP上で公表しているPediatricOncology Drug Approvals7)に記載の医薬品のうち、2000年から2022年に小児適応を取得した40品目の情報を調査対象とした。
日本における当該品目の承認状況や小児適応取得状況は、主に独立行政法人医薬品医療機器総合機構(PMDA)で公表している新医薬品の承認品目一覧や審査報告書、インタビューフォーム等から収集・分析した。
米国が含まれる臨床試験情報については、米国国立衛生研究所(NIH)等によって運営されている臨床試験登録システム(ClinicalTrials.gov)を用い、日本単独臨床情報についてはPMDA公表の審査報告書で確認した。
開発担い手の製薬大手はEvaluate Pharmaを参照し、小児適応取得時にグループ連結売上高が50億米ドル以上の企業とした。
日本未承認とは成人を含め新規有効成分の承認を得ていない品目を示し、日本適応外とは承認を得てはいるものの小児向けの適応を承認取得していない品目を示す。日本における承認および小児適応の有無は2023年3月末時点での情報を用いた。
3. 米国の小児がん用医薬品状況
3-1. 米国小児がん用医薬品の日本小児適応取得状況
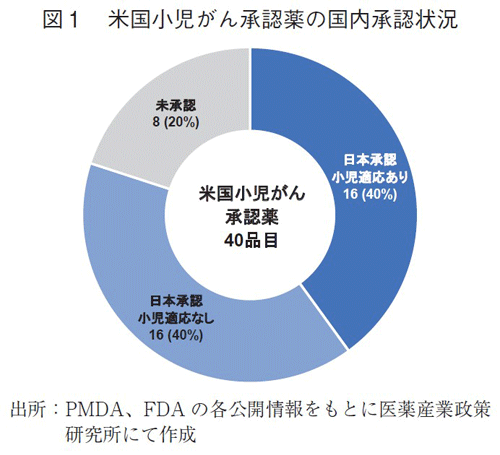
米国で小児適応を有する抗がん剤は2000年から2022年に40品目承認されていた。この40品目について日本での承認および小児適応状況を調査した(図1)。日本で承認されておりかつ小児の適応を取得している品目が16品目(40%)、日本で承認はされているものの小児の適応を取得していない品目が16品目(40%)、日本では未承認(2023年3月末時点)の品目が8品目(20%)であった。即ち、米国で小児がん用に承認されている40品目のうち、日本ではその60%にあたる24品目が小児適応を取得していないことが確認された。
3-2. 米国開発担い手分類別比較
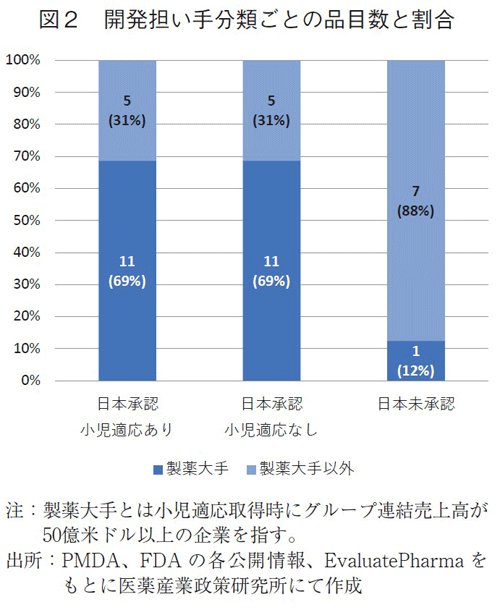
米国で小児適応を持つ40品目について米国での開発担い手を確認した(図2)。日本で小児適応のある16品目のうち、米国での開発担い手が製薬大手であった品目が11品目(69%)、製薬大手以外が5品目(31%)であった。
また、日本で承認されているが小児適応がない16品目では、製薬大手が11品目(69%)、製薬大手以外が5品目(31%)であった。
一方、日本未承認薬8品目では、製薬大手が1品目(12%)、製薬大手以外が7品目(88%)であった。
日本において承認されているが小児適応のない品目では製薬大手企業の開発品が多く、日本未承認の品目の大半は製薬大手以外の開発品であった。
3-3. ピボタル試験への日本組入れ状況
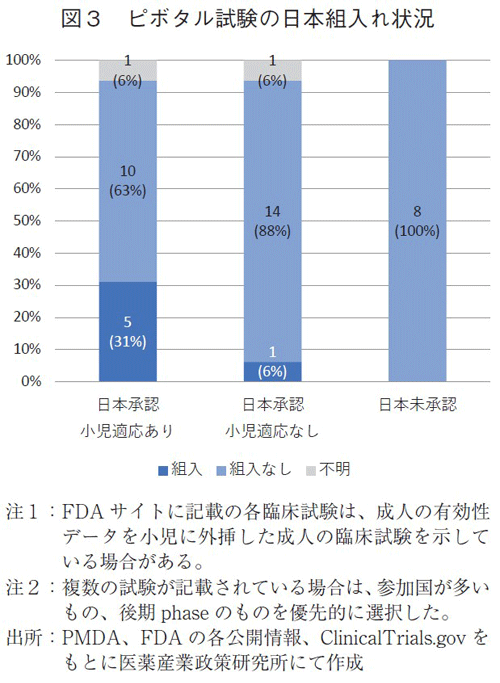
日本で抗がん剤が未承認となる要因として国際共同治験への日本地域組入れがないことが影響していることを政策研ニュースNo.66において報告している3)。小児適応取得においても関連性を探るべく、Pediatric Oncology Drug Approvalsに記載されている、承認の根拠となった臨床試験(ピボタル試験)での日本の組入れ状況を調べた(図3)。なお、日本からの組み入れ患者年齢について成人あるいは小児の別は調査に及んでいない。
日本で小児適応のある16品目のうち、日本が治験に組み入れられていた品目が5品目(31%)、組入れられていなかった品目が10品目(63%)、不明が1品目(6%)であった。一方、日本未承認と日本承認小児適応なしのほとんどが日本組み入れのない品目であり、日本未承認は8品目すべて、日本承認小児適応なしの品目は14品目(88%)が日本組み入れなしであった。
日本で小児適応を取得した品目においては、31%で国際共同治験に日本が組み入れられていることが確認されたが、興味深いことに日本が組み入れられていなくとも承認が取得されている品目が存在した。小児適応の承認取得においては、国際共同治験への参画以外のデータパッケージの活用もありうることが示唆された。
すなわち、日本未承認や小児適応なしの品目では、日本組み入れがほとんどないことが明らかとなり、少なくとも国際共同治験への日本地域の参画は承認取得のアプローチとして有用であることが示唆された。
4. 日本未承認・適応外薬の増加
4-1. 近年における米国小児適応取得品目の急増と日米差拡大
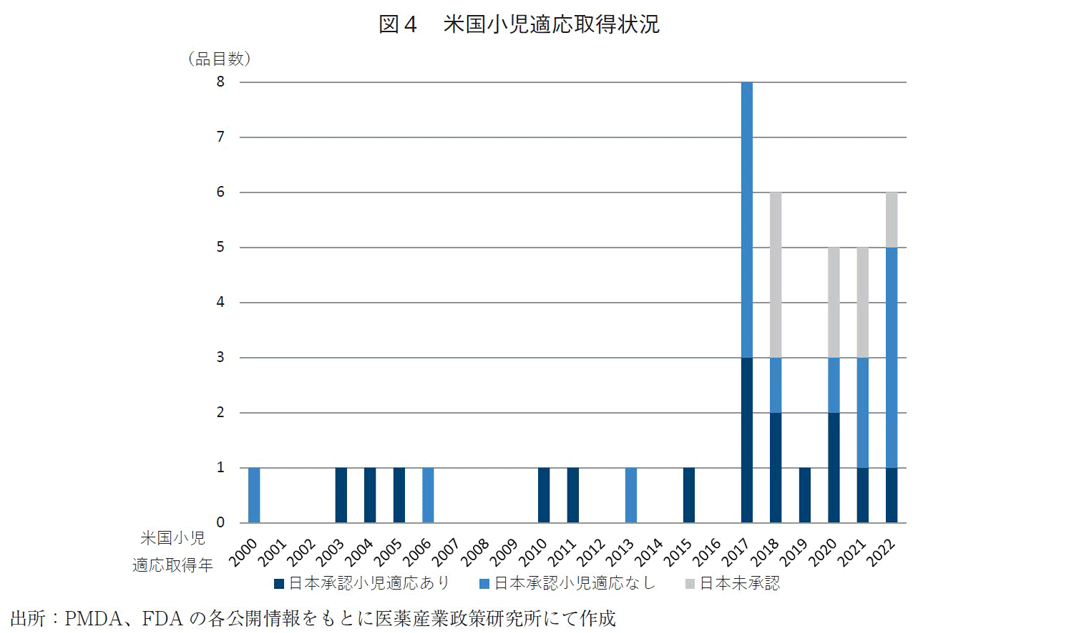
2000年以降の米国の抗がん剤のうち小児適応取得の品目数推移を分析した(図4)。小児適応取得年とその品目数を調査したところ、2000年から2016年の小児適応取得品目は9品目であるのに対し、2017年以降に小児適応取得したものが31品目(78%)あり、急増していた。
2017年以降の品目の日本承認状況をみると、日本で小児適応を取得した品目もある一方、2023年3月末時点にて日本で小児適応なしあるいは日本未承認薬が21品目(全体の53%)あり、多くの品目は日本で使用できないことが示された。
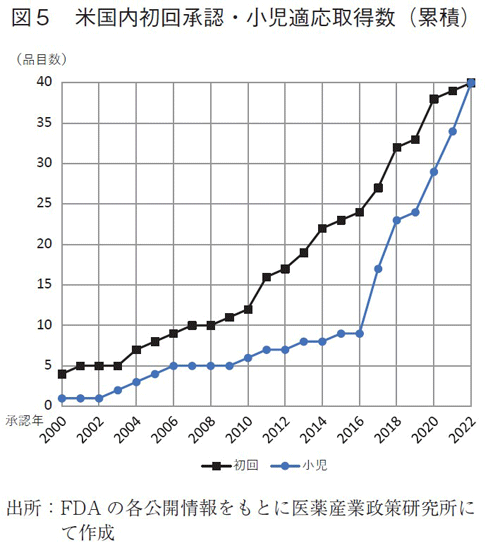
また、2017年以降の品目の変化について特徴を調査した。米国内での初回承認と小児適応取得数の推移を辿ったところ、初回承認の進度は従前とさほど変化なく着実に増加している中で小児適応取得数が急増していることがわかった(図5)。
さらに、日米の小児がん適応取得数の累積品目数を図6に示す。2017年以降の米国での承認品目が大きく増加したことに対し、日本の小児適応取得品目数では大きな増加は見られなかった。その結果として日米間の累積品目数の差が、2016年では3品目のみであったところ、2022年では24品目と拡大し、近年は日本で小児がんの未承認・適応外薬が増加していることが確認された。
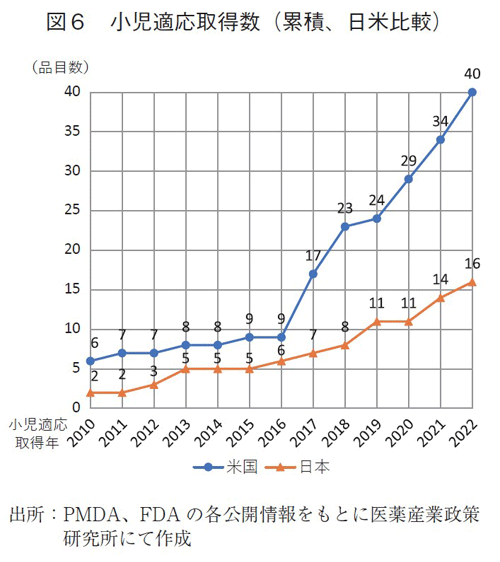
すなわち、2017年以降の小児適応取得における日米差拡大は、抗がん剤全体の承認に状況変化があったものではなく、米国における小児適応取得の状況変化に要因があったことが示唆された。
4-2. 初回適応取得か追加適応取得か
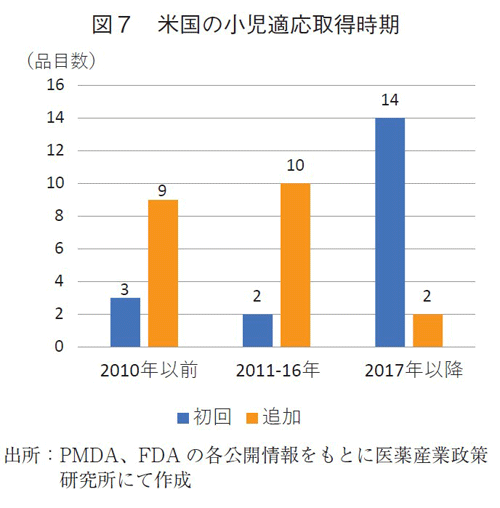
米国での承認品目につき、小児適応の取得タイミングを調査した(図7)。小児適応を初回承認時に取っているか、追加適応として初回承認後に取っているかに分類し、時期別(2010年以前、2011-2016年、2017以降)で集計した。
2016年以前には成人向けの抗がん剤として開発し、初回承認を得た後に小児適応を追加取得していた医薬品が多かった(9/12品目、10/12品目)。
一方、2017年以降はその比率が逆転し、初回承認時に小児適応を取得しているものが多く、16品目中14品目が初回時の取得であった。14品目のうち、成人患者にあまり使用されない、主に小児向けの抗がん剤は1品目のみであった。
5. 日本で小児適応のある品目の特徴
5-1. 国内小児適応取得時の臨床試験
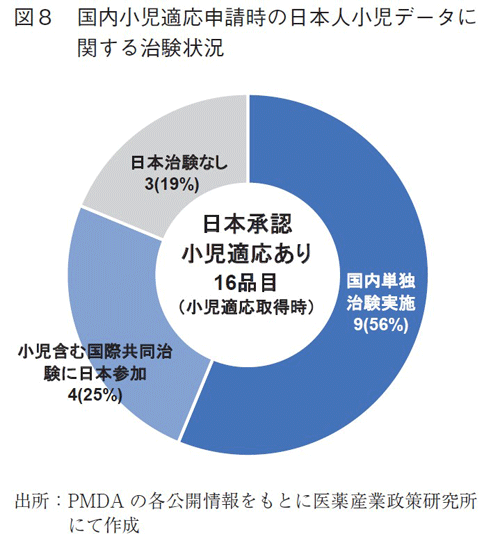
小児適応の取得における国際共同治験の位置づけを把握するため、日本承認小児適応ありの16品目について日本の小児適応取得時の臨床試験状況をPMDAの審査報告書から調査した(図8)。特に、日本人の小児が臨床試験に登録されているかどうかに着目して調査した結果、小児を含めた形で国内単独治験を国際共同治験とは別に実施していたものが9品目、小児患者を含んだ国際共同治験に日本が参加していたものが4品目確認された。さらに残り3品目のうち、1品目は主に海外データを用いた公知申請によるものであり、残り2品目は、申請時に日本人の小児データがなくとも、外国人小児データからの外挿等で承認取得していた8)。
すなわち、多くの場合で国際共同治験とは別に、日本人の小児データを取得するために、小児を対象とした国内単独の臨床試験を実施していることが明らかとなった。
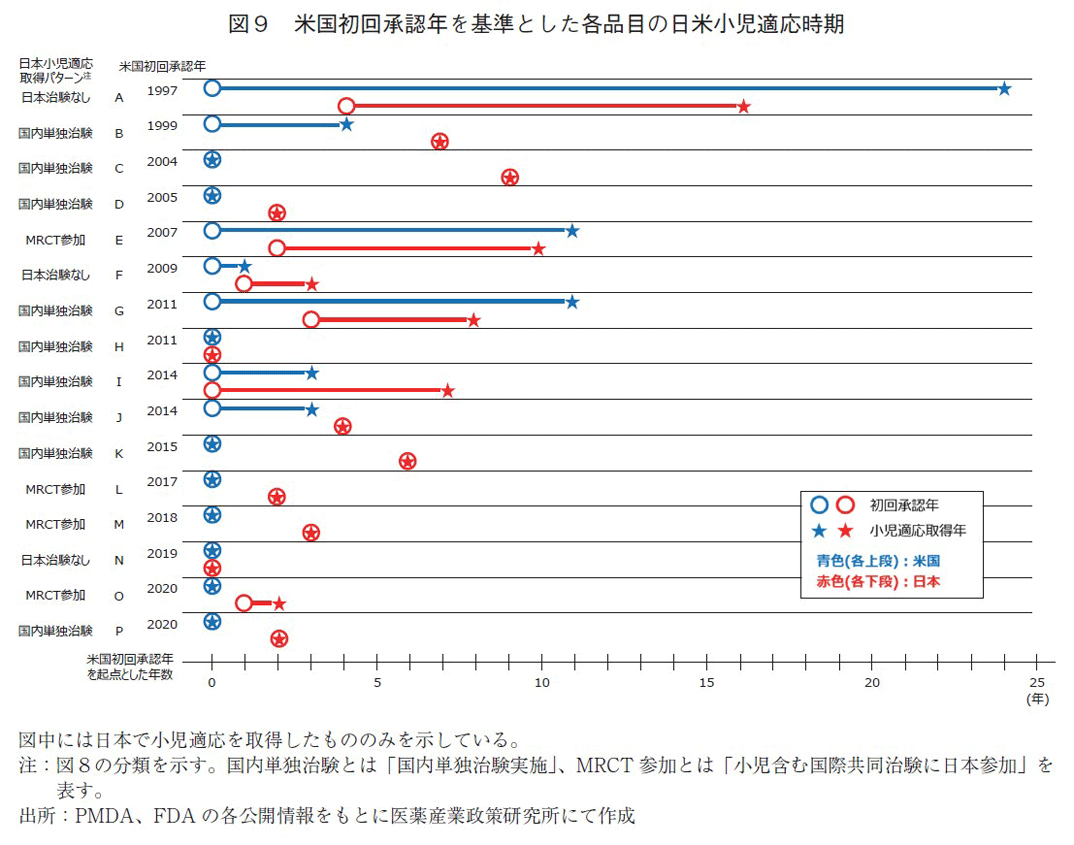
5-2. 米国初回承認から日本小児適応取得までの期間
日本承認で小児適応を取得した品目について、その承認時期の遅延有無を確認した(図9)。日米の両国にて小児適応が取得された品目は16品目あったが、それら全ての米国初回承認年を調査すると1997年から2020年であった。
各品目の承認適応取得パターンは複数あり、米国で初回から小児適応までの期間が長い品目(パターン1:A、E、G)、日本で初回から小児適応までの期間が長い品目(パターン2:A、E、I)などがあった。また、米国初回承認と日本初回承認の間に期間を要しているもの(パターン3:B、C)は、日本で初回と小児が同時であっても、米国からは遅れていた。
年代別に見てみると、近年では米国にて初回承認時に小児適応を取得する品目も多く、これらは比較的日本でも初回に小児適応を取っており、2017年以降では米国からの遅れも2年程度の品目であった(4品目/5品目)。すなわち、日本で小児適応が米国に遅延なく取得されるためには、米国で初回承認時に小児適応を取ること、日本でも初回承認時に小児適応を取ることが、極めて重要であることが示唆された。なお、本分析は既に日本で小児適応を取得した品目での結果となり、図に記載のない未承認・適応外の品目において今後小児適応を取得する可能性もある。また、各品目でデータ評価期間が揃っておらず、古い品目と新しい品目とを比較するには適していない。
6. 考察とまとめ
これまでを要約すると、米国で2000年以降適応を取得した小児がん用医薬品の60%は日本で未承認・適応外薬(2023年3月末時点)となっていた。詳細にはその3分の1が未承認であり3分の2が適応外薬であった。また、日本未承認薬となっている品目の担い手は、製薬大手以外の企業が多かったが、日本適応外薬となっている品目の担い手は、製薬企業も多く、大きい規模の企業でも日本での小児適応取得がされていなかった。さらには、2017年以降、米国の小児適応が初回承認時に取得され、品目数が急増した。そのため、日米の小児適応品目数の差が3品目から24品目に広がり抗がん剤の小児適応のドラッグ・ラグが拡大した。
まず、米国での2017年の変化について薬事規制の観点から考察を加えたい。抗がん剤の小児適応は、対象患者数も少ないこと、適応取得においては成人適応に追加して、臨床試験や製剤開発などの追加投資が必要となることが想定される。小児適応の開発が後回しとなる傾向は否めず、行政においても開発促進策が講じられてきた9、10)。米国においては2002年にインセンティブ(6か月の独占販売権、優先審査)を与えるBest Pharmaceuticalsfor Children Act(BPCA)11)、2003年に小児開発を義務付けるPediatric Research EquityAct(PREA)12)が制定された。また2012年には、Creating Hope Act13)により、譲渡可能な優先審査バウチャー(Priority Review Vouchers:PRV)がインセンティブとして付与されることが可能となっている。2017年には、Research to AccelerateCures and Equity(RACE)for Children Actが制定された14)。RACE for Children Actでは新たに、FDAが制定した小児がんでも関連性の高いとする分子標的リストに基づき、成人向けの当該分子標的抗がん剤を開発している企業に対し、小児試験計画を作成することが義務付けられた。2017年からの米国における小児適応品目数の増加の背景には、法制定から適応取得までの開発期間を考慮すると、ここに記した各種インセンティブと義務付けの両側面の開発促進策が影響した可能性がある。加えて、米国の小児適応の加速要因について、21世紀に入りがんゲノムと疾病との関係性解明といった科学の進歩、個別化医療の進展などの表れである可能性も考えられる。RACE for ChildrenActにおける分子標的リスト公表もその一端と捉えられる。
一方、日本においても開発促進策が講じられ、承認後の再審査期間の延長(10年を超えない範囲)とその明確化を企図した見直し(令和2年)、薬価の小児加算(5-20%の引き上げ)などが挙げられる15)。また、特定用途医薬品指定制度16)も制定され、優先相談・優先審査などの小児の治療薬開発を促す施策が施されているが、米国に比べ未承認・適応外薬の品目が増え小児適応取得の品目数差が拡大する現状の中では、より効果的な施策が必要となることが想像に難くない。
小児適応の抗がん剤においては、そもそもその品目が日本では未承認となっている品目がある(8品目)ことが明らかとなった。3-2章で示した通り、日本未承認薬の多くは製薬大手以外が開発を担っていた。これは小児がんの未承認薬に限った事象というより、近年のドラッグ・ラグやドラッグ・ロス全般に見られる要因である3)。すなわち、新興企業(本稿では製薬大手以外に該当)にとって、日本の治験実施環境のハードルが高く、また、日本地域への投資にあたっては、日本の薬剤市場の魅力度が低い可能性がある。実際の企業行動や思考については、公開情報からの推察には限界があり、個社毎の実態聞き取り調査等より深い精査が必要とされる。
また、日本では承認されているものの、小児の適応が取得されていないもの(16品目)も多くあったが、これらの開発担い手の多くは製薬企業大手であることが示された。製薬企業大手では、日本国内に開発機能・販売機能やノウハウを有しているものの、開発着手されていない現状では、小児開発の追加投資を回収する見込みが立たないことが要因として想定される。また、抗がん剤の領域では、遺伝子変異を標的とした分子標的薬が主流になりつつある。ある種希少フラグメント化しており、投資回収の困難度が高まっていると同時に、データ収集の困難さが増している。小児開発への投資負担を軽減するような行政による財政上の支援と、海外含む既存データの外挿で承認を与え、承認後の国内実臨床データをもって再審査する事例を増やすなどの薬事制度支援、両面からの強力な開発支援が大いに期待される。
本稿からは論点が多少ずれるが、小児がんは成人のがんと比較して疾病治療以外にも考慮すべき点がある。例えば長期入院に伴う就学支援の課題などがあり、国立がん研究センターでは「小児がん就学の相談対応の手引き」17)を発行している。あるいは、晩期合併症18)、妊孕性温存19)等の課題もある。医療関係者のみならず家族の看護体制など総合的な援助策が必要であり、社会全体として取り組む課題と考え、製薬に携わる者として日本での治療選択肢を増やし、救える・治せる・より良く生きられる子供が増えることを願うばかりである。
-
1)医薬産業政策研究所「ドラッグ・ラグ:国内未承認薬の状況とその特徴」政策研ニュースNo.63(2021年7月)
-
2)医薬産業政策研究所「ドラッグ・ラグ:未承認薬は日本のアンメット・メディカル・ニーズに応えうるか?」政策研ニュースNo.66(2022年7月)
-
3)医薬産業政策研究所「ドラッグ・ラグ:なぜ、未承認薬が増えているのか?」政策研ニュースNo.66(2022年7月)
-
4)医薬産業政策研究所「ドラッグ・ラグ:日本と欧州の未承認薬状況の比較」政策研ニュースNo.67(2022年11月)
-
5)医薬産業政策研究所「新薬の国際普及の計量分析:米国承認新薬の日欧承認に注目して」政策研ニュースNo.67(2022年11月)
-
6)
-
7)
-
8)
-
9)「欧米における小児医薬品開発の取組み」中里有希 ファルマシア、2022、58(3)、207-211.
-
10)“Changing incentives to ACCELERATE drug development for paediatric cancer” T. de Rojas, P. Kearns, P. Blanc, J. Skolnik, E. Fox, L. Knox, R. Rousseau, F. Doz, N. Bird, A. J. Pearson, G. Vassal, Cancer Medicine., 2023;12:8825-8837.
-
11)
-
12)
-
13)
-
14)
-
15)「小児医薬品開発における行政の取組み」堀内大士 ファルマシア、2022、58(3)、201-206.
-
16)
-
17)
-
18)
-
19)