Points of View 医薬品の開発期間の調査 ~モダリティ及び疾患の違いは重要か~
医薬産業政策研究所 主任研究員 高橋洋介
はじめに
政策研ニュースNo.66において、医薬品の研究開発における「成功確率」に関する分析を行った1)。研究開発の生産性を分析する上では、「成功確率」だけでなく、「研究開発期間」や「研究開発コスト」が重要な要素となり、これらを総合して考察することで1つの新薬を上市するために必要と想定される研究開発投資額を見積もることも可能となる。研究開発期間は、新薬上市までの機会費用に影響するだけでなく、上市後の特許保護期間中の独占販売期間などにも影響するため、研究開発投資の現在価値ベースの収益性を大きく左右する。本稿においては、医薬品データベースに収載されている情報をもとに「研究開発期間」に関して分析を行うこととした。
方法と結果
本研究では、2つの手法によって開発期間の評価を行った。1つの目の手法は、各開発候補品の適応疾患毎に、各フェイズにおける期間を算出する方法である。2つ目の手法は、各開発候補品(NME)に焦点を当て、何らかの適応疾患で最初に臨床試験を開始した日から、何らかの適応疾患(試験開始時とは異なる適応疾患でも可とする)で最初に承認を得るまでの期間を算出する方法である。前者の手法では、各フェイズの平均的な開発期間の把握や、適応疾患(領域)毎の平均的な開発期間の把握等に有用で、医薬品の研究開発期間の分析時に汎用される手法であるが、一つの開発候補品(NME)に着目した開発期間を捉えることが出来ていないという制約がある。この点を考慮したのが2つ目の手法であり、適応疾患の変更などにより開発過程で紆余曲折を経た期間もすべて組み込み、NME毎に開発に要した期間を算出している。
本稿の一連の調査研究においては、医薬品データベースであるEvaluate Pharmaを用いた。まず手法1によりPhase1、Phase2、Phase3の各期間及び申請から承認に要する審査期間(Filedとして図示)を調査した。具体的には、Developmental RiskモジュールのTimeline分析を用いて、調査日時点において収載されている全データを対象とした。本データベースでは、各フェイズの期間を当該フェイズ開始日から次段階のフェイズの開始日として定義されており2)、臨床試験の期間等ではないことに留意する必要がある。審査期間は、FDAへの申請後にFDAにて承認されるまでの期間として定義されており、COVID-19ワクチンで見られたような緊急使用許可(EUA;Emergency Use Authorization)を得るまでの期間ではなく、正式承認されるまでの期間として算出されている。
なお、データベースに収載されている開発期間に関わるデータは、最終的に承認に至った医薬品だけではなく、開発中の薬剤や開発中止となった薬剤であっても、次相へと進んだ早期フェイズのデータについては集計対象とされている。例えば、Phase2実施後に開発中止が決定された開発品Xがあった場合、開発品XのPhase1の期間はデータベースに収載され、Phase2以降の期間は収載されない。
開発期間の中央値などの要約統計量を解析する上では、比較的近年の状況を反映する目的と、データの信頼性を高めるために一定のサンプル数を確保する目的から、各フェイズの開始日を表1の通りに限定して集計している。データベースに収載されている情報は、ClinicalTrial.govの登録情報など、製薬企業等から公開された情報に基づいているため、必ずしも全開発品の開発期間の情報が網羅されているわけではない点に留意する必要がある。
手法2では、FDAで承認された新薬(NME)に関して、承認年毎に分類した後に、それらNMEが最初にPhase2を開始した年月を調査し、その差から「Phase2開始から承認までに要した期間(NME毎)」を算出した。なお、Phase1開始日から承認までに要した期間の算出も試みたが、承認薬に限定した場合にはPhase1開始日が明確でない割合が高く、分析に足るサンプル数が確保できないと判断し、この解析は行わなかった。
各フェイズにおける開発期間
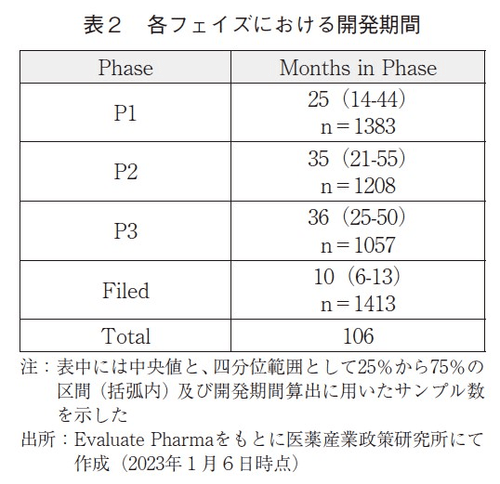
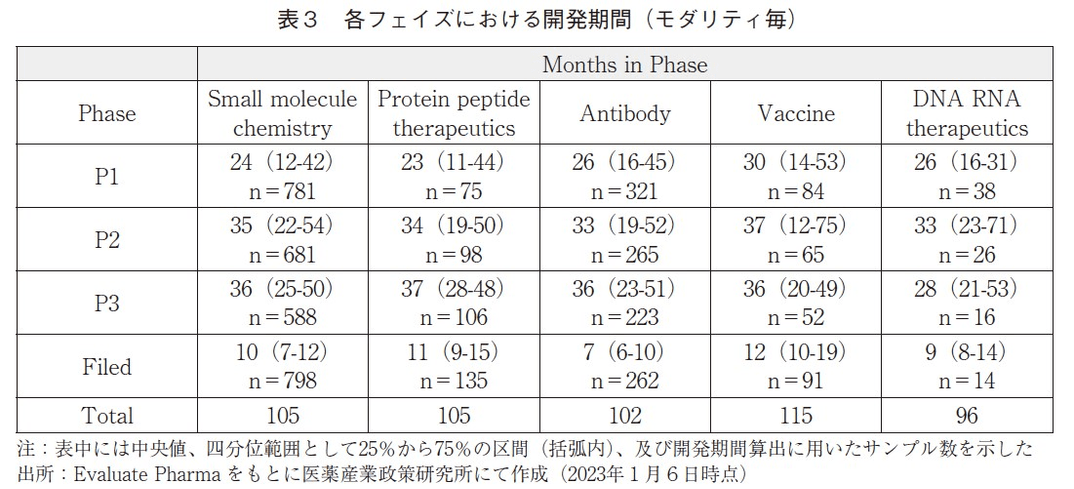
各フェイズにおいて要した開発期間を、データベースに収載されている全データから算出し、中央値及び四分位範囲として25%から75%の区間と、これら数値算出に用いたサンプル数を表2に示した。また、モダリティ毎に分類した開発期間を算出し、同様に表3に示した3)。表3でのモダリティ分類4)は、低分子(Small molecule)、組換えタンパク(Protein peptide therapeutics)、抗体(Antibody)、ワクチン類(Vaccine)5)、核酸(DNARNA therapeutics)であり、その他のモダリティについてはサンプル数が少ないと判断しデータ集計対象からは除外した。
その結果、Phase1の期間の中央値は25か月、Phase2は35か月、Phase3は36か月、審査期間は10か月であり、これら期間の合算値は106か月(8年10か月)であった。ただし、開発期間は開発品毎のバラつきが非常に大きく、中央値近辺に収束するのではなく、25%から75%の四分位範囲はPhase1で14~44か月、Phase2で21~55か月、Phase3で25~50か月、Filedで6~13か月と、特に早期段階のフェイズほど比較的大きな広がりを持っていた。新規作用機序や新規モダリティの開発品であった場合に、Phase1試験では安全性に最大限配慮して低投与量から慎重に投与量を増大させていくなど、開発品の特性に応じた試験プロトコールが組まれるケースが多いことや、次相の試験プロトコールを最適化するために追加試験が組まれるケースがあることなどが、早期段階のフェイズほど比較的大きな広がりを持っている要因ではないかと考えられる。
各フェイズにおける開発期間(モダリティ別)
モダリティ毎の分析結果(表3)では、集計対象の半数程度を占める低分子の開発期間は、全医薬品を対象とした開発期間(表2)と概ね同程度で、Phase1の期間の中央値は24か月、Phase2は35か月、Phase3は36か月、審査期間は10か月であり、これら期間の合算値は105か月であった。また、組換えタンパクや抗体の開発期間も全医薬品を対象に算出した開発期間と概ね同程度であり、開発期間の合算値はそれぞれ105か月及び102か月であった。ワクチン類の開発期間はやや長く、開発期間の合算値は115か月であり、一方で核酸医薬の開発期間はやや短く、開発期間の合算値は96か月であった。
ワクチン類の開発期間に関して掘り下げて分析をする。表3の通り、ワクチン類の開発期間は、Phase1、Phase2、Phase3及び審査期間の中央値がそれぞれ30か月、37か月、36か月、12か月であり、これら期間の合算値は115か月という結果であった。さらに、COVID-19パンデミック前の2019年以前のデータに限定して集計しなおした場合には、それぞれ30か月、42.5か月、40か月、12か月の開発期間となり、これら期間の合算値は124.5か月となった。つまり2019年以前では、ワクチン類の開発期間は他モダリティに比べて特に長い傾向にあったことが分かる。にもかかわらず、2019年12月に中国で原因不明の肺炎(後のCOVID-19)の発生が報告されていて以降6)、様々な画期的ワクチンが創製され世界中の人々を救うことに繋がったが、これらが過去に類を見ないほどのスピードで実用化に至った点において特に注目すべき事例である7)8)。例えば、FDAはPfizer-BioNTech社のワクチンを2020年12月11日に緊急使用許可9)、2021年8月23日に正式承認10)しており、Moderna社のワクチンを2020年12月18日に緊急使用許可11)、2022年1月31日に正式承認12)している。COVID-19パンデミック下において、ワクチンが短期間で次々と実用化された背景には、新型コロナウイルスの病原性や感染性・伝播性の高さなどその脅威の大きさに基づき、国家的な政策などの様々な緊急措置が取られたこと、多数の被験者を速やかに集められたことなどが短期間での開発を可能とした一つの要因と考えられるだろう。ただし、そのような政策的・疾患特異的な要因だけではなく、イノベーションの進展が開発期間を短縮した点も注目に値する。mRNAワクチンのように、基礎研究の積み重ねによってもたらされた新技術により短期間で高い有効性を発揮するワクチン創製が可能になったことや、デジタルトランスフォーメーションの進展に基づき、ワクチン設計期間の短縮だけでなく、臨床においてリアルタイムでデータが収集され速やかに効果の解析が行われたことや積極的にリアルワールドデータが活用されたことなども重要な要因であるだろう。このような観点に立つと、COVID-19パンデミックで得た(さらに、今後継続的に蓄積していく)知識や経験は、その他のワクチン開発を効率化させる可能性もあり、将来的にワクチン類の開発期間が短縮していくことが期待される。
核酸医薬においては、Phase3期間が中央値で28か月とやや短い傾向にあった。期間算出に用いたサンプル数が16と他モダリティに比べて少ないため、考察の確からしさに留意する必要があるものの、この16品目のうち9品目がExpedited approval(迅速承認)13)の経路を経てFDAで承認されている点が特徴的である。核酸医薬であるから開発期間が短くなるというよりは、低分子医薬などの従来型モダリティでは治療が困難であった疾患に対して核酸医薬による創薬が試みられ、そのようなアンメットニーズの高い疾患では特に一刻も早く患者さんに新薬を届ける必要があることから、臨床試験の評価項目においてサロゲートエンドポイントを採用するなどの対応が行われ、開発期間が短縮化される方向に繋がったのではないだろうか。
各フェイズにおける開発期間(疾患領域別)
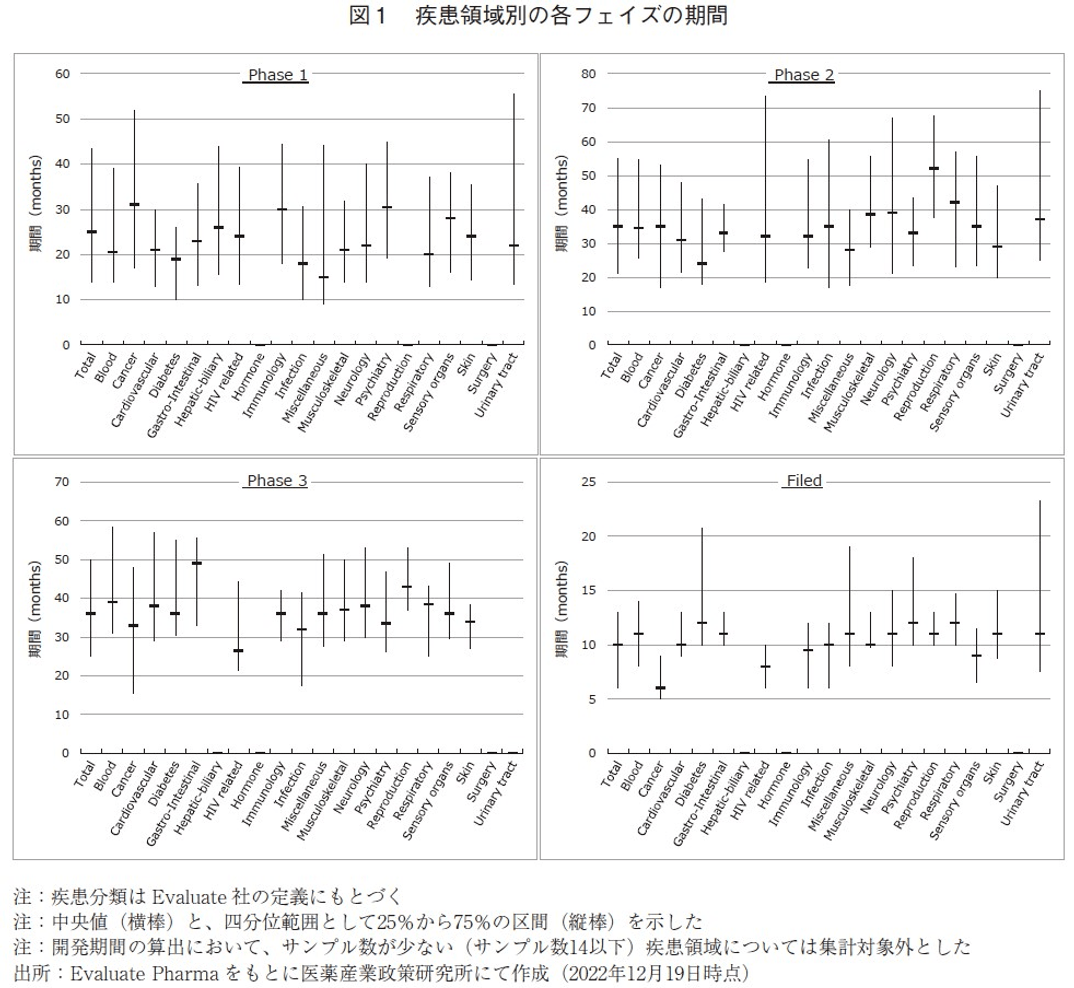
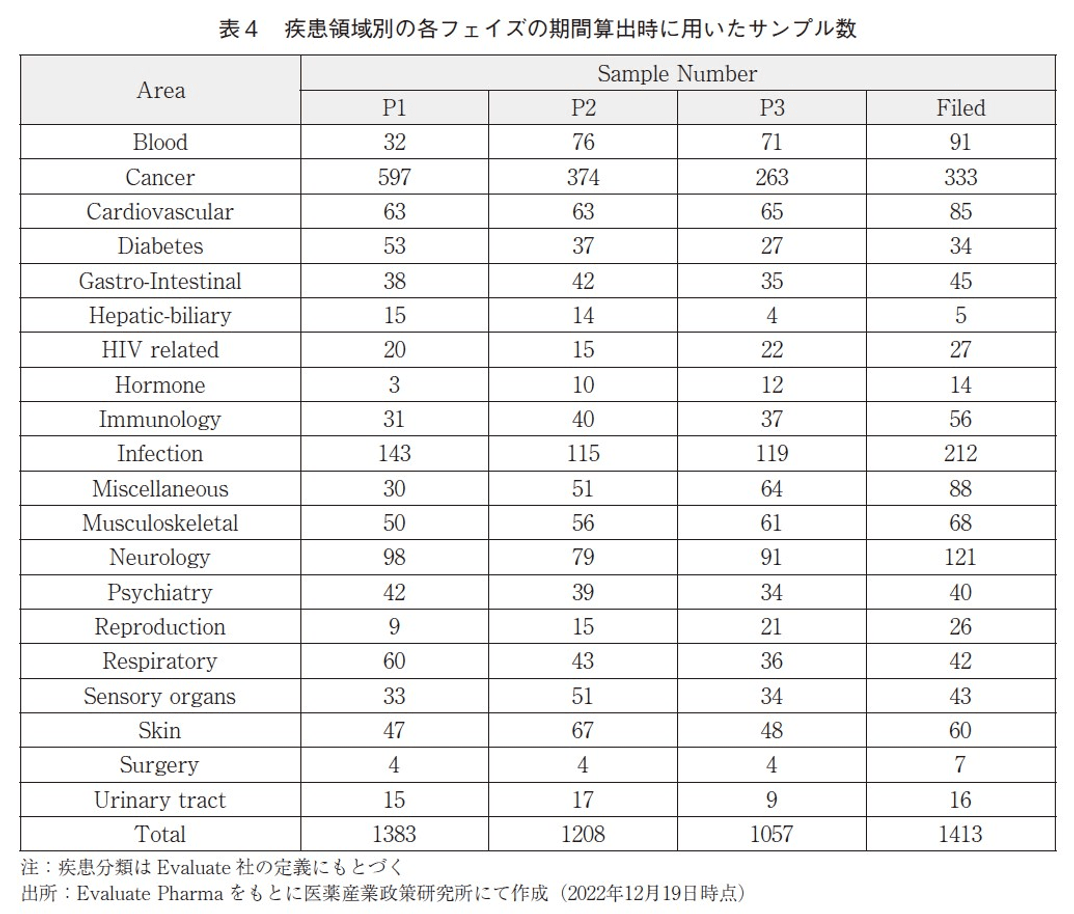
次いで、疾患領域別に各フェイズの期間を調査した。疾患領域の分類方法に関してはEvaluatePharma上での分類基準を用いた14)。同一疾患領域であっても開発品毎に開発期間の大きなバラつきが存在していたため、図1では、各フェイズにおける開発期間の中央値と、四分位範囲として25%から75%の区間を示した。また、疾患領域毎の開発期間算出に際しては、一定のサンプル数(各フェイズにおいて15以上)が確保出来るものに限定し、サンプル数は表4に示した。
Phase1の期間に関して、多くの疾患領域で共通して概ね中央値20~30か月で次相に移行しており、疾患領域による大きな差は認められなかった。ただしCancer領域では、やや開発期間が長い傾向にあった。これはCancer領域では患者でのPhase1試験が行われることが多く、安全性や薬物動態特性だけでなく、有効性に関する評価も行われることがあり、期間が長期化していることが考えられる。
Phase2の期間に関して、多くの疾患領域で概ね30~40か月で次相に移行していることが確認出来たが、Reproductionの領域ではやや開発期間が長いという結果であった。Reproduction領域には、子宮筋腫、子宮内膜症、不妊、早産、更年期障害などの疾患が含まれている。これら疾患に対する開発品で実際にPhase2に要した期間を精査すると、他疾患領域での開発期間の中央値である30~40か月を大きく超える開発期間を要した事例が散見された(個別データは割愛)。これら疾患の中には、未だアンメットニーズの高い疾患が多く含まれており、開発に成功した先行医薬品が多くないという特徴がある。そのために臨床試験計画の立案時に前例に倣うことが困難であったことなどが、開発期間の長さに起因していると想定出来たが、これ以上の精緻な分析は困難であった。ただし、本領域では開発期間算出に用いたサンプル数が評価対象下限の15と同数で、他の疾患領域と比べてもサンプル数が少ないことから、このことが本結果に影響を与えている可能性も否定できない。
Phase3の期間に関しては、概ね30~40か月で次段階(申請)に移行しているが、GastrointestinalとReproductionの領域でやや長い期間を要していた。Gastrointestinalには様々な疾患が含まれており、その中では潰瘍性大腸炎やクローン病、便秘などを適応疾患とする開発品で開発期間が長い傾向にあったが、精緻な考察は困難であった(詳細なデータは割愛)。
審査期間は、概ね10か月前後で大きな違いは認められなかったが、唯一Cancerの領域でやや短く6か月程度という結果であった。今回の分析に用いた事例の中では、Cancerの領域では審査期間の算出に用いた事例のうち約2/3(332件中204件)でExpedited approval(迅速承認)のタグがついており、Cancer以外の領域では約1/4(1081件中255件)でのみExpedited approval(迅速承認)のタグがついていた。この差が審査期間の差に現れている可能性が考えられる。
Phase2開始から承認までに要した期間(NME毎)
前述の「各フェイズにおける開発期間」の調査では、あらゆる開発品を対象として網羅的に集計した後、実際に要した開発期間をフェイズ毎に算出し、それら各フェイズの開発期間を合算する方法によって、Phase1から承認までの医薬品の開発期間などを評価している。この方法では、各フェイズに分解して平均的な期間を評価可能というメリットがあるが、一つの開発品の研究開発プロセスにおいては、必ずしも各フェイズが直列的に実施されるわけではなく、各開発品の実質的な開発期間を正しく評価出来ない(≒短く見積もる)可能性がある。
上記のような懸念点が想定されることから、一つ一つの開発品に着目して、「Phase2開始から承認までに要した期間(NME毎)」を分析した。また、近年の状況について把握することを目的として、FDAでの承認年毎に分類して評価を行った。
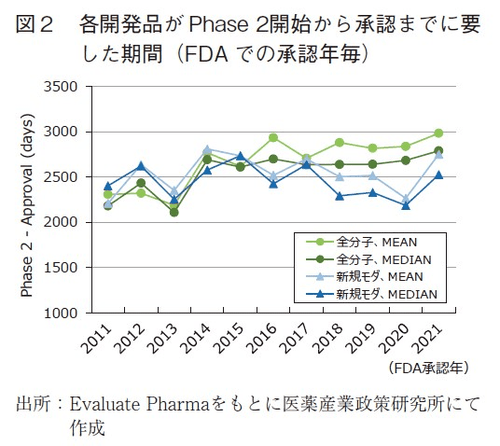
図2では、Phase2開始日及びFDA承認日の情報が得られる全開発品での分析結果を示し、加えてその中から新規モダリティ15)のみを抽出した分析結果を示した。開発期間算出方法の違いについては図3に例示した。「Phase2開始から承認までに要した期間(NME毎)」の中央値は、近年では概ね2,600日~2,700日(7年2か月~7年5か月)前後で安定していた。新規モダリティに限定すると、この期間の中央値は2,300日~2,500日前後であり、やや短いという結果であった。表2の結果をもとに合算して算出すると、Phase2から申請までに要する期間は81か月(6年9か月)となる。両手法で算出される期間の差は5~8か月であり、「Phase2開始から承認までに要した期間(NME毎)」の方が長いという結果であった。この数字の差の要因については、分析対象としている母集団の違いに基づく可能性は排除できないものの、図3に示したような例(開発段階で適応疾患を変更するなどの紆余曲折を経てた例)が一定数存在していることも一因となっているだろう。
あとがき
本稿では、データベースを用いて医薬品の開発期間を調査・分析することで、研究開発の生産性(1つの新薬を上市するために必要と想定される研究開発投資の大きさ)の一要素を評価することを試みた。研究開発の生産性を検討する上で重要な要素として、主に「成功確率」、「研究開発期間」、「研究開発コスト」が挙げられる。政策研ニュースNo.66においては、「成功確率」について調査を行った1)。成功確率に関しては、モダリティ毎や疾患領域毎で比較した場合には、大小様々な数値が算出され大きな差異が認められ、成功確率を正確に推測・把握することが研究開発の生産性を可視化する上で重要であると考えられた。一方で、本稿で「研究開発期間」を分析した結果では、一部の事例を除いてモダリティ毎や疾患領域毎で大きな差異は認められないという結果であった。つまり、過去事例を元にすれば、開発期間を大まかに概算することは可能と考えられた。
ただし、この結論にはいくつか留意点がある。分析上の限界と今後の課題について、以下に記述する。一点目は、開発期間の中央値を分析する限りは大きな差異は認められないものの、個別の開発品に目を移した場合には大きなバラツキが認められており、ミクロな視点で分析する場合には適していないということである。二点目は、各フェイズの期間の合算値が、個々の開発品の開発期間に必ずしも合致しないという点である。図3で示したように一つの開発品に関して、適応疾患を変更するなどして複数の同一相の試験が実施される場合などでは、実際のトータルの開発期間は各フェイズの期間の合算値よりも長期化しているケースが存在する。三点目として、各フェイズの期間を「当該フェイズの開始日から次フェイズの開始日までの期間」と定義しているため、あるフェイズで開発中止となり次相へと進まなかった場合には、当該フェイズの期間は集計対象外となっている点である。つまり開発失敗事例については、開発期間の推計は不十分ということである。今回の結果を元に、創薬に成功したケースと失敗したケースで開発期間に差があったかを分析することは難しい。四点目として、本稿ではデータベースに収載されている情報を用いて分析しているため、各製薬企業等から公表されている情報が中心となってしまい、臨床試験着手以前の基礎研究や非臨床試験の期間を算出出来ておらず、創薬の全工程における開発期間は算出出来ていない点である。
今回の分析結果から、現在の製薬産業の姿を把握する上で有用な知見を提供することは可能であるものの、前述の通り様々なデータ上の制約が存在しており、十分な分析結果を得られたとは言い難い。データベース等では調査不可能な情報を得るため、政策研では過去に製薬協加盟企業を対象としたアンケート調査を行い、製薬産業の研究開発の生産性を可視化する研究を行っている17)。しかし本調査は2000年から2008年の期間を対象とした分析である。現在の製薬産業の姿は調査当時から大きく変化していることが推察され、今一度新たに同様の手法による調査研究を行い、今回の調査研究において残された課題に対して取り組むことも検討してみたい。
-
1)医薬産業政策研究所、「創薬の成功確率分析 -臨床試験に焦点を当てて-」、政策研ニュース No.66(2022年7月)
-
2)例えば、Phase1期間はPhase1開始日からPhase2の開始日として定義される。また、Phase1開始日やPhase2開始日は、原則としてClinicalTrial.govにおいて「start date」として記載されている日付として定義されている。
-
3)モダリティ毎の開発期間算出においては、一定のサンプル数(各フェイズにおいて二桁以上)が確保出来るものに限定した。
-
4)モダリティ分類は、Evaluate Pharmaにおける技術分類を用いた。ただし、Antibodyに関してはMonoclonal antibody及びRecombinant antibodyの合算とした。
低分子医薬:Small molecule chemistry、抗体医薬:Antibody、組換えタンパク:Protein peptide therapeutics、ワクチン類:Vaccine、核酸医薬:DNA RNA therapeutics -
5)ワクチン類(Vaccine)について、Evaluate Pharmaの分類基準ではワクチンとして用いられる様々なモダリティの総計である。例えば、組換えタンパクであっても、ワクチン用途で用いられる場合にはProtein peptide therapeuticsではなくVaccineに優先的に分類される。
-
6)Zhu N, Zhang D, Wang W, et al. A Novel Coronavirus from Patients with Pneumonia in China, 2019、N Engl J Med. 2020 Feb 20;382(8): 727-733.
-
7)医薬産業政策研究所、「日米欧の新薬承認状況と審査期間の比較 -COVID-19ワクチンの事例も踏まえた日本の課題-」、政策研ニュース No.64(2021年11月)
-
8)医薬産業政策研究所、「感染症予防ワクチンの創製について」、政策研ニュース No.65(2022年3月)
-
9)
-
10)
-
11)
-
12)
-
13)Evaluate Pharmaでは、日米欧においてPriority Review、Fast Track、Breakthrough therapy、Qualified Infectious Disease Product (QIDP)、Accelerated Approval、RMAT (Regenerative Medicine Advanced Therapy)、PRIME(PRIority MEdicines) Program、Sakigakeのいずれかに該当した開発品に対して、Expedited approvalのタグが付与されている。
-
14)Evaluate Pharmaでは、疾患領域をBlood、Cancer、Cardiovascular、Diabetes、Gastro-Intestinal、Hepatic-biliary、HIV related、Hormone、Immunology、Infection、Miscellaneous、Musculoskeletal、Neurology、Psychiatry、Reproduction、Respiratory、Sensory organs、Skin、Surgery、Urinary tractに分類しており、これらすべてを合算したものをTotalとした。
-
15)Evaluate PharmaにおいてBiotechnologyとして分類されるモダリティを新規モダリティとして抽出しており、抗体などのバイオ医薬品の他、化学合成によって製造される核酸医薬などもここに含まれる
-
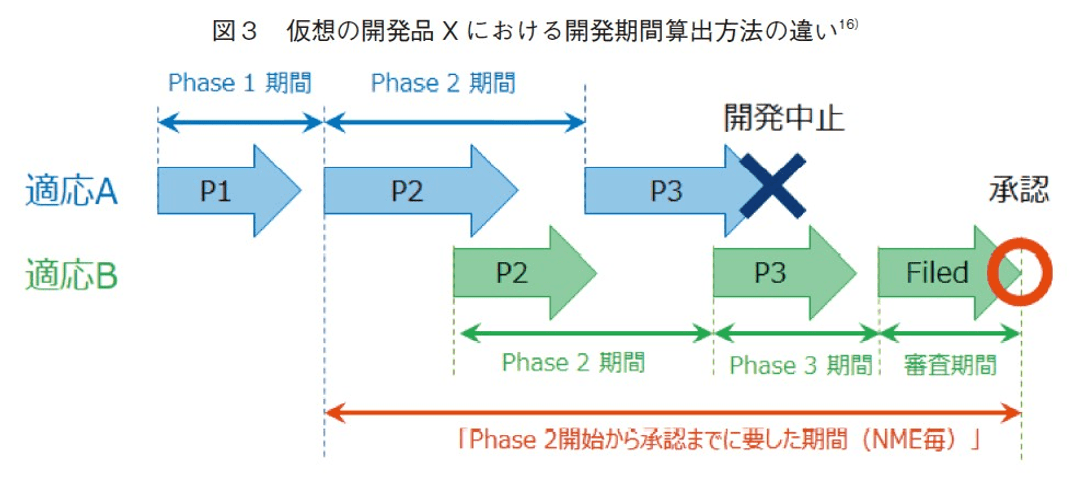
16)仮想の開発品Xの開発過程を示した。適応疾患Aで開発が先行し、適応疾患Bでも後追いで開発が進められた。適応疾患AではPhase3で思わしくない結果を得て開発中止を決定し、適応疾患Bでは承認までたどり着いている。このケースでは「各フェイズにおける開発期間」を算出する際には、Phase2期間として2つの期間を得ることになり、それぞれが区別して集計される。一方で「Phase2開始から承認までに要した期間(NME毎)」を算出する際には、適応疾患AのPhase2開始日から適応疾患Bでの承認日までの期間を得ることになる。
-
17)医薬産業政策研究所、「医薬品開発の期間と費用 アンケートによる実態調査」、リサーチペーパー・シリーズ No.59(2013年7月)