Topics
新薬の国際普及の計量分析:米国承認新薬の日欧承認に注目して
医薬産業政策研究所 所長、東京経済大学教授 長岡貞男
医薬産業政策研究所 客員研究員、学習院大学教授 西村淳一
医薬産業政策研究所 主任研究員 吉田昌生
1. 背景と目的
新薬が国際的に普及していくことは、患者が利用可能な医薬品の多様性を高めると共に、新薬創出からの利益を高めることで新薬の研究開発も促すので、イノベーションの促進にとって極めて重要である。
新薬の国際普及については、Cockburn, Lanjouw, and Schankerman(2016)2)等の先行研究はあるが、その変動の原因について実証的に解明されていない問題も未だ多い。また、日本では近年、ドラッグ・ラグの再燃が懸念されており、その原因を分析していく上でも重要なテーマとなっている。既刊の政策研ニュース(第63号及び第66号)において、2010年代の後半に日本の新薬未承認率が拡大傾向にあることを指摘し3)、またその原因の候補についても徹底した分析がなされている。本ニュースでは吉田(2022)4)が、米国での承認品目について日本と欧州の承認の比較分析を新たに行っている。
本稿はこれらの分析を補完して、新薬の国際普及に与える各種要因の重要性を計量的に分析することを試みている。新薬の特徴(米国FDAのブレークスルーセラピー指定あるいはオーファンドラッグ指定)5)、新薬開発企業の特性(新興企業かどうか)、及び知的財産保護期間が新薬の国際普及に如何に重要か、及びこれらの日欧間の差を分析する。このため、2010年から2021年までに米国で承認された新薬(新有効成分含有医薬品)について、日米欧各国の承認情報(2022年初頭まで)とIQVIAの上市データと接続を行い、これを利用した計量分析を行った。
本稿の構成は以下の通りである。次節で先行研究を述べ、続いて本稿の分析に利用したデータの構築方法を説明し、次に上市のモデルとその推計モデルを説明し、推計結果を述べる。分析では、米国が先行上市した新薬についてCOX比例ハザードモデルで分析し、更に米国が日本あるいは欧州と同年承認したサンプルを加えて、同年承認について線形確率モデルで分析している。
2. 先行研究
新薬の世界的な普及について、Cockburn, Lanjouw, and Schankerman(2016)が最も代表的な研究となっている。グローバルに最初に上市された新薬がその後各国に普及する過程を分析し、ある国で新薬が上市されるかどうかに価格規制が負の影響を与え、また、特許権の保護の強さ(物質特許や保護期間の長さ等)が正の影響を与え、新薬が創製された後でも、その各国への普及には臨床試験へのインセンティブが重要であることを示している(1983~2002年における76か国で上市された672の新有効成分含有医薬品を対象)。推定によれば、強い価格規制によって、その国における新薬の上市確率は15%減少し、(予想される上市ラグの期待値よりも)25%程度上回る上市ラグをもたらす。また、物質特許制度がある国における上市ラグは55%短くなる。更に、特許制度と価格規制には相互依存関係があり、強い価格規制のもとでは特許保護の影響が相殺されることが示されている6)。
Gaessler and Wagner(2022)7)はデータ保護期間に着目して、独占保護期間の影響を分析している。新薬の特許を対象に欧州特許庁(EPO)への異議申し立てが新薬開発途中に成立した新薬を分析対象としており、その結果として、当該新薬の市場での独占保護期間が減少した程度を測定することで開発進展と日米欧における承認確率への影響を線形確率モデルで評価している。推定の結果、1年間の市場での独占保護期間の喪失は平均的にみて4.9%ポイントの承認確率の減少となっていた。特に、臨床試験の初期に異議申し立ての結果が出されることの影響が大きく、開発当事者が大企業であるほど承認確率は減少することが見出された。
また日本における新薬開発遅延について、今井・成川(2022)8)が分析をしており、2010年から2020年に米国および欧州双方で承認された新有効成分含有医薬品を対象として、日本における承認がそれよりも遅い、あるいは2020年末の時点で未承認である新薬について、Cox回帰分析を行っている。その結果、類似薬が存在しない新薬、欧米で承認を取得した企業に日本法人がない新薬、オリジネーター企業と日本での承認取得企業が一致していない新薬等では、日本で開発が遅延することを示している。
本稿では、新薬の特徴(ブレークスルー指定あるいはオーファン指定)、新薬開発企業の特性(新興企業かどうか)そして知的財産保護期間が、日欧における当該新薬の承認率をどのように予測しているかを、日本と欧州の差を含めて定量的に分析していること、そして新薬の国際普及において重要になっている国際共同治験の成果だと考えられる同年承認の分析も行っていることが新たな貢献となっている。
3. データ構築
本稿の分析では主に2種類のデータを利用している。まず、米国で承認された新薬の日欧での承認実績のデータであり、本ニュースの吉田(2022)で利用されている、医薬産業政策研究所が構築しているデータである。分析対象サンプルは米国(FDA)が2010-2021年に承認した481新有効成分含有医薬品9)である。欧州と日本の承認については直近まで把握しており、2022年に承認された新薬も存在する。米国市場は新薬の革新性を高く評価する市場10)で世界の新薬販売額の4割程度を占め、また医薬産業政策研究所の定点観測によると11)、米国に基盤をおく企業は世界の売上高上位品目創出の過半を占めており、米国市場は非常に重要な地位を占めていると考えられる。このため、本稿では米国で承認された新薬に注目する。
もう一つのデータソースはIQVIAのPricing Insightsである。本データは、各国ごとに上市されている医薬品の上市日、価格、知的財産保護期間等のデータを収載している。本研究では成分名(Molecules)と米国での製品名によって、承認データをIQVIAと接続した。成分名や製品名に表記の差(表記ゆれ)がある場合には目視で確認した12)。我々のPricing Insightsのデータは2019年の初期までのデータをカバーしており、これを利用した分析では2018年までに米国で承認された新薬を対象としている。
4. 上市のモデルと分析方法
4.1 臨床開発投資の意思決定
新薬がある国で承認され上市されるためには、企業が当該国での審査に足る臨床開発投資を行うことが必要であり、そのためには、当該国で新薬を上市することによって得られる利益が臨床開発投資を上回ることが必要である13)。
上市からの期待利益 > 臨床開発投資の期待コスト (1)
新薬の革新性を評価する程度(薬価等のインセンティブ)や市場の規模(患者数)が高まれば、上市後の期待利益は高くなり、特許保護期間が長ければ、新薬上市から利益獲得可能な期間は長くなる。これらの要因は上記の(1)式が満たされる確率を高め、新薬を上市しやすくする。
市場や臨床試験コストの状況は時間の経過によって変化するので、ある時点で(1)式が成立しなくても(失敗の確率が高く、上市からの期待利益が小さい)、将来の時点では成立するようになる可能性もある。また、(1)式が成立していても、臨床試験コストはサンクコストになるので、待つことで不確実性が減少する場合には、企業にとっては臨床試験の実施を遅らす方が有利になる場合もある14)。
他方で、特許権による独占実施期間は新薬が発明され特許を取得した時点で決まっており、臨床試験の実施が遅れれば、特許権の保護による独占実施が可能な期間は短くなる。この効果は、企業に早期の臨床試験実施を促す。但し、臨床開発投資がデータ保護期間によって保護されている場合には、その期間は審査承認時点からカウントされるので、特許権による保護と異なって早期の臨床試験を促す効果は無い。
このように、臨床開発投資はリスクのある先行投資であり、もし新薬保有企業がリスク資金調達の壁に直面していれば、(1)式が成立しても臨床試験は実施されない。また新興企業は各国での販売基盤が弱く、外部企業との販促分野での業務連携が必要となり、臨床試験の実施が遅れ結果的に期待利益が小さくなる可能性もある。
以下では、新薬の革新性、新薬の開発企業の類型、知的財産保護期間に着目して、これらが新薬の日欧での承認確率を説明する程度、及び欧州と日本の差を統計的に分析する。それぞれについて、以下の予測をすることができる。
- ①革新性が高い新薬は上市後の期待利益が高いが、同時に臨床開発の費用や不確実性も高い可能性がある。米国先行承認の場合には臨床開発の不確実性は低く、その費用も下がると考えられ、一方で革新性の高い医薬品の米国における高薬価が日欧へも部分的に反映される可能性が増すため、期待利益への効果が優越し、日欧で承認される確率は高いと考えられる。
- ②新興企業が新薬保有者である場合、資金制約等によって日欧で承認される確率は低下する。
- ③特許権の残存保護期間が長ければ、日欧で当該新薬が承認される確率は高い。
また、これらの効果の日欧の差について、日本に比べて欧州の方が市場の規模が大きく、独、英等では日本より革新性を評価する程度が大きいと予想されるので、
- ④革新性の効果や特許権の残存保護期間の効果は、欧州での効果がより大きく、逆に新興企業の負の効果は小さいと予想される。
以下では、こうした予想が成立するかどうかを検証する。
4.2 分析方法
米国で承認された新薬には、米国が日欧いずれに対しても先行して承認した場合と、日本あるいは欧州と同年に承認した場合15)がある。企業は先ず同年承認を得るかどうかを選択するが、以下ではそれを線形確率モデルで分析する。同年承認ではなく、米国が先行している場合の日欧での承認確率をCox比例ハザードモデルで分析する。いずれも米国で承認された新薬が日米で承認される過程を確率的なイベントとして把握する。
Cox比例ハザードモデルでは、承認イベントが単位時間当たりに生起する確率(hazard rate)を左右する要因は、経過時間によらず同じ割合で(比例的に)作用することを仮定したモデルである。経過時間によらず比例的に作用するという条件はかなり強い仮定であるが、他方でこのモデルでは、イベントが発生していない打ち切りデータも含め、イベントの発生についての利用可能なデータを全て整合的に取り入れて推計できる点で優れている16)。また、Cox比例ハザードモデルではイベントの発生確率について特定の分布の仮定をしない利点もある。データの制約上、本推計における経過時間は年として把握する。
ある新薬iがある国jにおいて経過年tの時点内に承認される確率hは、以下のように表記される。xijは承認確率に影響を与える、医薬品単位あるいは承認国毎の属性であり(複数の要因が存在する場合はベクトルとなる)、推計される係数βの強さで承認確率に影響をする。λ(t)はxijが全てゼロである場合の経過時間毎の承認確率の時間分布である。誤差項を省略して
h(t│xij)=λ(t)exp(βxij) (2)
対数をとると、
lnh(t│xij)=lnλ(t)+βxij
となり、属性xijの1単位の変化が係数βに等しいだけハザードレートの対数を変化させることが分かる。
現実には国際共同治験などによって、米国と日本あるいは欧州で同年に承認される場合が少なくない。ハザードモデルでは経過時間0ではイベントが起きる確率はゼロとなり、米国承認との同年の時点での承認を分析の対象とすることは困難である。この制約を回避するために、本稿では米国承認と同年の承認が日本あるいは欧州でなされる確率(H(xij))を被説明変数とした推計モデルを以下の線形回帰モデルによって分析する(誤差項省略)。
EH(t│xij)=δxij+α (3)
この分析では、承認データの打ち切りの影響をコントロールすることが特に重要であり、日欧での未承認新薬を含めて分析対象サンプルとするとともに、FDAの承認年ダミーで打ち切りの影響をコントロールする。
いずれのモデルにおいてもxijは外生変数であり、(2)式及び(3)式の誤差項(ランダムな要因)とは独立であること、並びに2つの式の誤差項は独立であることが前提とされている。しかし実際には、我々が把握できず、かつランダムでない要因が存在するかもしれない。我々は、医薬品のATC分類(疾患分類と作用機序を示す分類)等をコントロール変数として導入することである程度、その要因の影響を緩和しているが、それがなお存在するので以下の分析は因果推論の推計ではない。各変数は相関するが欠落している要因の影響も捉えていることに留意する必要がある。また、各新薬について日欧2つの観測値があり、その誤差項の相関をコントロールするために標準誤差の測定ではクラスタリングをしている。
5. データの概観
以下の表1に米国先行、米国と同年、日欧先行に分けた場合の新薬の特徴、及び日欧の承認率を示している(米国では全て承認されている)。280の新薬で米国承認が先行し、日欧では未承認か米国より遅く承認した。132の新薬では欧州あるいは日本と同年承認となっている。これらの新薬がブレークスルー指定される確率は同年承認の場合が少し大きいが米国先行とほぼ等しく、それぞれ27%と24%である。オーファン指定される確率も両者でほぼ等しい。それぞれにおいて、日本の承認比率は欧州承認比率よりも大幅に低い。米国が先行している場合、日本では2022年初頭時点で34%、欧州では52%が承認されている。また、米国と日欧いずれかが同年承認の場合、欧州ではほぼ全て(96%)が承認されているが、日本では67%にとどまる。すなわち、欧州の方が米国承認と同年承認となることが多く、また、そうでない場合も日本より早く承認されている。欧州の場合は、同年承認の新薬の数(132×0.96=127)と米国に遅れて承認した新薬の数(280×0.52=146)はかなり近く、国際共同治験による同年承認の重要性の高まりを示唆している。同年承認の場合、臨床試験企業が新興企業である確率は大幅に低く、米国先行の場合は45%、同年承認の場合は20%である。
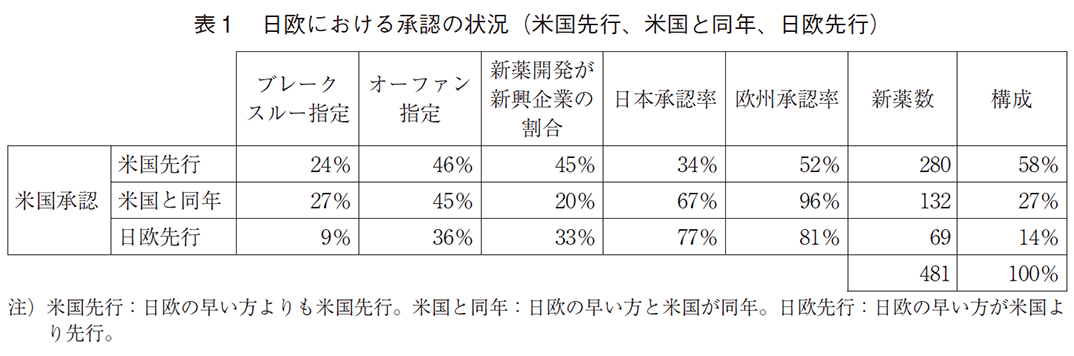
表1の最後の列が日欧いずれかが先行の場合で、全体の14%存在する。ブレークスルー指定の割合は米国先行あるいは米国と同年の場合と比較して半分未満である。
6. 米国先行承認の新薬についてのCox比例ハザードモデルによる分析
2010年から2021年の間に、米国で日本及び欧州よりも承認年が早い新薬について(日欧で未承認の新薬を含む)、日本と欧州の直近(2022年初頭を含む)までの審査状況を対象として、(2)式を推計した結果を表2に示す。米国承認年を起点として、欧州の承認及び日本の承認が生ずる確率(対数)を推計した結果であり、表2にその確率に影響を与える因子(説明変数)のリストも示している。全てのモデルで米国の承認年ダミー(コホート年ダミー)を導入し、臨床試験方法の変化等の日欧に共通する経時的な環境変化をコントロールしている。各変数の係数の推計値はハザード比ではなく、ハザードレート(hazard rate、承認が各年で起きる確率)の対数値をどれだけ変化させるか(ハザードレートがほぼ何%変化するか)を示している17)。ハザードレートが高ければ承認までのラグは短く、また、一定期間に承認される確率は高くなる。
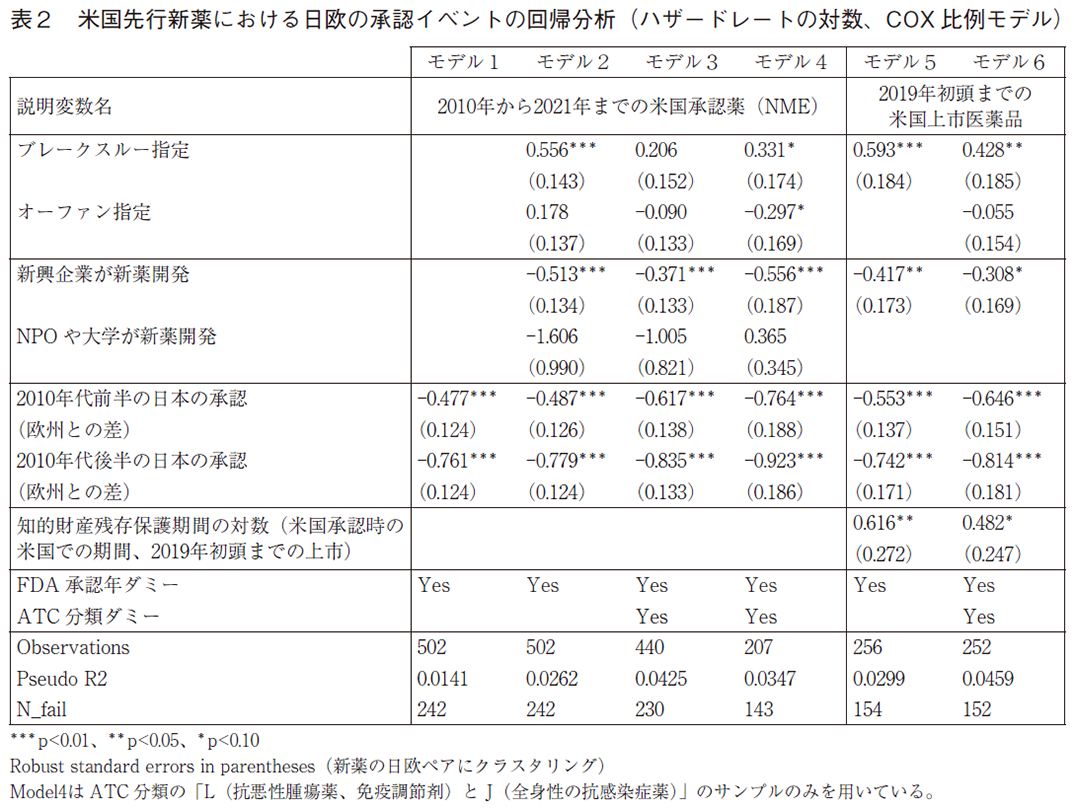
モデル1は最も単純な推計モデルであり、欧州と比較した日本の承認のハザードレートの対数値の差(推計値はマイナスなので減少度合い)を2010年代の前半の米国承認のサンプル(2010-2014)と後半承認のサンプル(2015-2021)に分けて評価している。推計結果によれば、2010年代の前半に承認された新薬と後半に承認された新薬のいずれの場合でも、日本の承認確率は欧州と比較して統計的に有意に低い。2010年代の前半の新薬で約48%ハザードレートが低く(式(2)のβに相当する係数の推計値が-0.477)、後半の新薬でも約76%低い。前半と後半ではハザードレートを計測している期間の長さが異なり、その大小を直接比較することはできない。7節で確認するように、日本の承認プロセスと比較して、欧州では米国での承認後の早期に承認が集中してなされることを多分に反映している18)。
日本の承認確率が欧州の承認確率と比較して大幅に低い点は、モデル2及び3において、新薬の特徴や新薬開発企業の特性などを導入し(モデル2)、またATC大分類ダミーをコントロールしても(モデル3)変わらない19)。なお、革新性等の要因が日欧に作用する大きさが異なる可能性は以下の日欧別の効果の推計で分析する。
次に、新薬が米国でブレークスルー指定あるいはオーファン指定をされているかどうかの影響を分析する。モデル2によれば、ブレークスルー指定は日欧の承認確率の約56%の上昇(式(2)のβに相当する係数の推計値が0.556)を伴っているが、ATC大分類ダミー変数を追加した推計(モデル3)では統計的な有意性が無くなるので、疾患分野の影響が大きいことが分かる(ブレークスルー指定が多い疾患で全般的な承認率が高い)。但し、ブレークスルー指定の頻度が高いL(抗悪性腫瘍薬、免疫調節剤)及びJ(全身性の抗感染症薬)の分野(新薬の約半数)に推計を限定した場合(モデル4)でも、ブレークスルー指定は有意であり(係数は33%で、5%有意に近い)、革新性が高い新薬は日欧に普及しやすい傾向があることがなお示唆される。またモデル5と6に示すように、米国における2019年初頭までの上市薬に限定したサンプルでは、ATC分類と知的財産保護の程度を追加した推計でも(モデル6)、ブレークスルー指定の係数は有意で40%の水準である。全体として、強いエビデンスではないものの、4.1節の予測①と整合的で革新性の高い新薬は日欧で早期に上市されやすい。
他方で、オーファン指定と日欧の承認率には有意な関係は無い。オーファン指定は患者の数が少ないことが指定要件の一つであり、この点は臨床開発投資にマイナスである。他方で新薬が既存の治療薬が無く満たされていない治療への需要を満たしている革新性も示し、またオーファンドラッグに特化したインセンティブも日欧で存在し、これらが相殺していると考えられる。但し、L及びJの分野では、推計された係数はマイナス30%と、統計的に有意に承認確率は低い。
次に臨床試験の開発企業が新興企業20)である場合の影響を見ると、全てのモデルで頑健に大きく負である。ATC分野をコントロールしたモデル3によれば、承認確率は37%低下し、L及びJの分野に特化した推計(モデル4)では56%低下である。この結果は新興企業が直面する資金制約の重要性を示唆しており、上記の4.1節の予測②と整合的である。NPOや大学がスポンサーである場合も大きな負の係数が観察されるが、統計的な有意性は無い(サンプルサイズが小さく、検出力が弱いことが要因だと考えられる)。
米国承認時の米国での知的財産残存保護期間(対数)の影響の推計値は、モデル5とATC大分類を追加的にコントロールしたモデル6で示されており、有意に正である。臨床開発投資を行うかどうかには特に知的財産残存保護期間が短い場合にクリティカルな問題となる可能性が高く、それを反映させて対数値を利用している。知的財産残存保護期間についての情報は2019年初頭までに米国で上市されている承認薬についてしか利用可能ではなく、これら二つのモデルは観測数が少ない(約半分となっている)。モデル(5)では、保護期間の長さが10%延びると、年間の承認確率を約6%高めることを示唆している。ATC分類をコントロールしても有意であり(モデル(6))、効果は約5%である21)。
以上の知的財産残存保護期間の推計は過小評価となっている可能性がある。推計では米国承認時での知的財産保護期間の残存期間を、日欧において企業が臨床試験に成功した場合に利用可能な残存期間を示す代理変数として利用しているが、これは特許権の保護についてのみ妥当である。特許権による保護の開始時点は日米欧共通であり、例えば米国での承認時点で残存期間が長ければ、日欧でも特許権による残存保護期間は長くなる22)。他方で、臨床試験投資の収益性を保護する制度としてのデータ保護(日本の再審査制度)期間の方が独占実施期間を決めている場合、それによる保護は各国承認時からカウントされるので、特許権の保護の場合のような保護期間の医薬品間変動の国際的連動性はない(各国で類型毎に該当医薬品で共通期間となる制度のため)。我々が使用するデータでは、データ保護がどの程度優勢であるかを把握していないので区別ができない。しかし、日米欧州で実際に上市された新薬のデータを利用した分析によれば、米国における知的財産残存保護期間の変動は日独における知的財産残存保護期間の変動を有意に説明している23)。
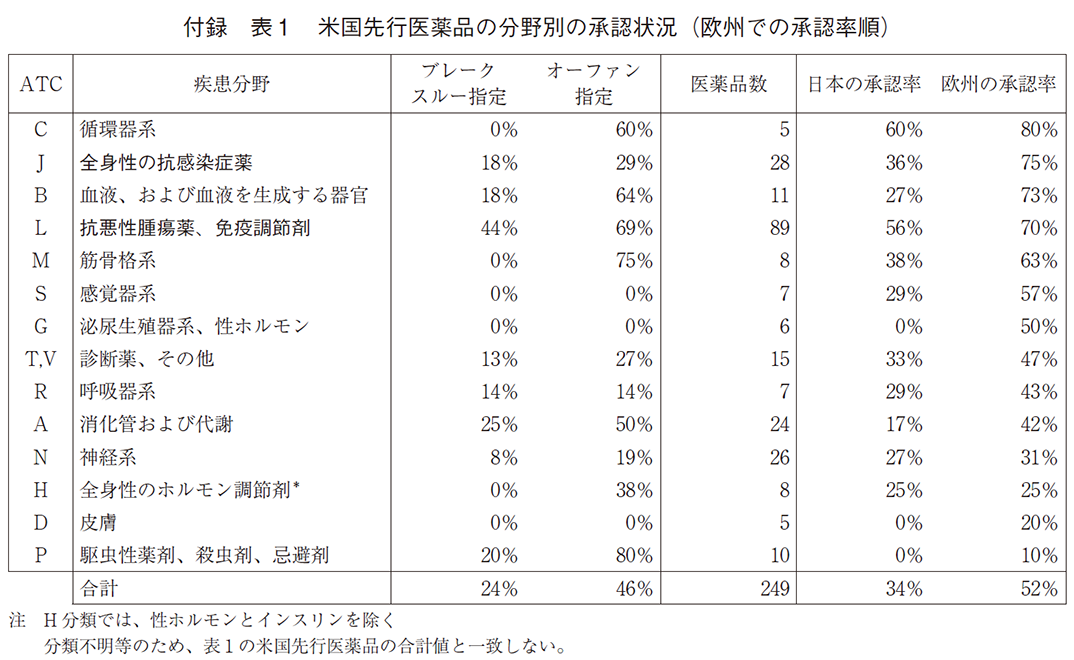
以下の表3は、新薬がブレークスルー指定をされているかどうか、開発企業が新興企業であるかどうか、そしてFDA承認時での米国での知的財産残存保護期間が長いかどうか(9年を越えるかどうか)によって、承認率がどの程度変化するかを、欧州と日本それぞれにおいて示している。新薬がブレークスルー指定をされていると日本でも欧州でも承認率は高く、開発企業が新興企業である場合に低い。ただ注目すべき点は、欧州では日本と比較して差が大幅に小さいことである(12%対26%)。知的財産残存保護期間が短い新薬の承認率は日欧共に低い。
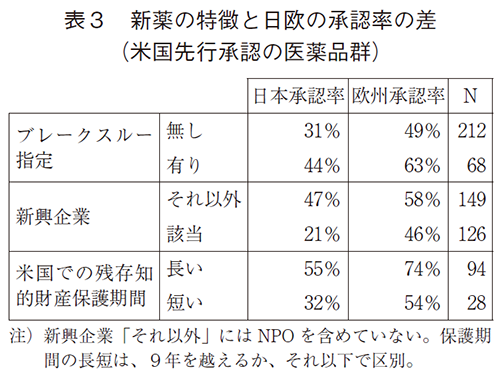
表3は、4.1節の予想④で述べたように、日欧で新薬の革新性等がその承認率に与える効果が異なる可能性を示しているが、以下の表4ではその統計的有意性を検証している。推計モデルは、表2の拡張であり、表3の3つの新薬の特徴が日欧で異なる影響を与えることを許容するように拡張している。データは表2と同じく日欧の承認イベントをプールしており、日欧でCOX比例ハザードモデルの基礎となるハザードの時間分布は同一としている(これによって日欧の比較が可能となる)。
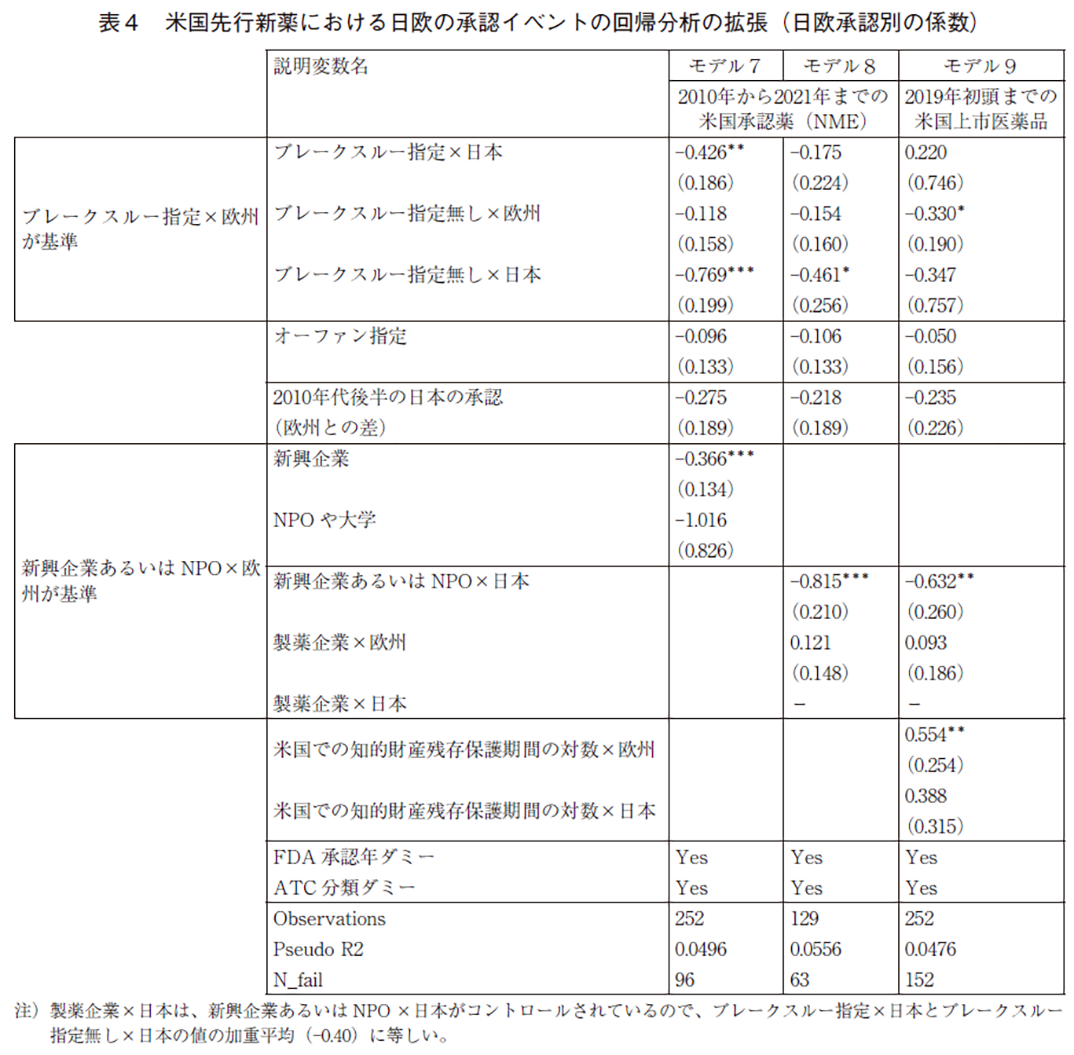
ブレークスルー指定の係数のみ日米差を許容した場合がモデル7、新興企業の影響も日米差を許容した場合がモデル8、知的財産残存保護期間にもそれを許容したのがモデル9である。ブレークスルー指定の新薬で、新興企業あるいはNPOが開発企業であり、かつ欧州での承認である場合を推計の基準としている。モデル(7)によれば、ブレークスルー指定の新薬の日本の承認確率は欧州よりも43%低い。但し、ブレークスルー指定が無い場合に、日本の新薬承認率はそれ以上に欧州より低い(ブレークスルー指定無し×欧州とブレークスルー指定無し×日本の係数の差は0.651)。したがって、革新性の正の影響は欧州でより大きいという、4.1の予想④は成立していない。
モデル(8)によれば、新興企業あるいはNPOであることは、欧州と比較して日本に非常に大きな負の影響を与えている。承認確率への影響はマイナス82%である。また同時に、ブレークスルー指定である場合の日本の承認率へのマイナスの影響はかなり小さくなり、統計的にも有意ではなくなる。更に、モデル(9)によると、知的財産残存保護期間については、欧州のみがプラスで有意であり、日本ではそれが長くなることが承認確率に与える有意な影響は観察されなかった(但し、差は有意ではない)。新興企業あるいはNPOが日本でのみ大きく負で有意であることは依然として成立する。
全体として、日欧の差についての4.1の予想④は、新興企業の影響と知的財産保護の効果については成立している。ただ、欧州では前者の負の影響は大幅に小さく、後者では欧州のみで有意にプラスであるものの、日本との差は統計的には有意ではない。したがって、日欧の承認確率の最も重要な差は、新興企業が開発企業である場合に日本での臨床開発投資が困難なことにあると考えられる。
7. 米国承認先行新薬の日欧における承認のダイナミクス
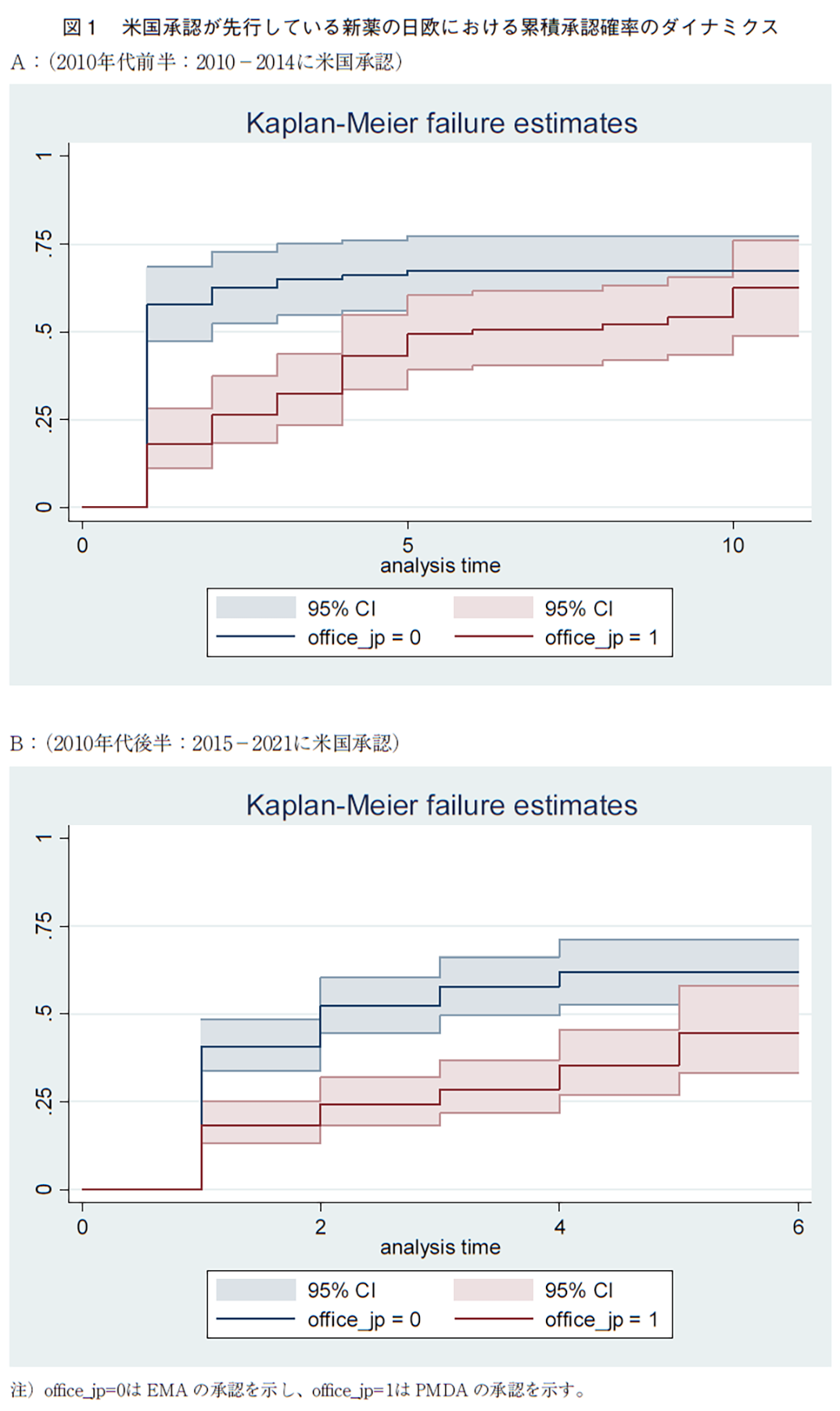
日欧において承認のダイナミクスは大きく異なる。図1は、以上で分析してきた米国承認が先行している新薬について、それが日欧で承認される確率が米国の承認年を起点とした経過年によってどのように変化するかを示している(Kaplan-Meier failure estimates)。パネルAが米国で2010年代前半(2010-2014)に承認された新薬であり、パネルBは2010年代後半(2015-2021)に承認された新薬である。これによれば、欧州では米国での先行承認の直後から高い水準で承認がなされ、前半では2年目、後半では3年目には米国の50%を越え、前半では5年目で75%を越えてフラットになる。これに対して、日本では立ち上がりが遅く、毎年ほぼ一定割合で承認が増えていき、米国の50%を越えるのは2010年代の前半で10年目である。図1では信頼区間も示されているが、我々が持っている比較的小数のサンプルでも5年目までは有意に欧州の承認確率の方が高い。このように、欧州と日本では承認の水準のみならず、経過時点によるその変動のパターンも異なるので、経過期間の長さによって、欧州と日本の平均承認確率の差は変化する(日欧で比例的にハザードレイトが異なるとの仮定は成立しない)ことに留意が必要である。
8. 米国と日欧の同年承認確率の分析
以下の表5は、日本と欧州がそれぞれ米国と同年に承認した新薬の頻度を集計している。母数は、米国が先行して承認した新薬及び米国が日本あるいは欧州と同年に承認した新薬である(日米欧州が同年の事例は無かった)。母数は表1で米国先行承認と米国との同年承認のケースの和であり、日欧承認先行の場合は除いている。
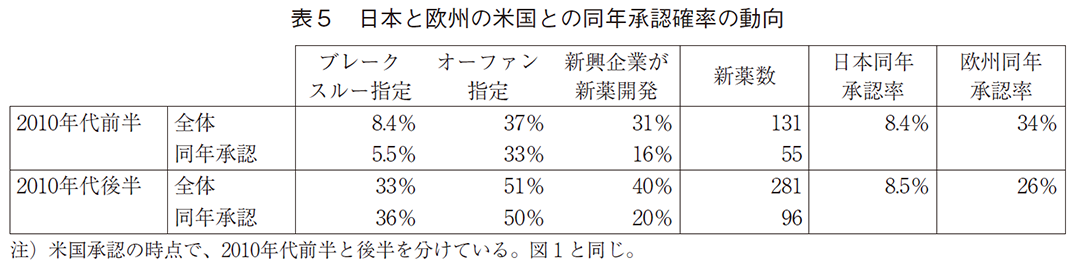
日本では2010年代の前半も後半も、同年承認の確率は約8.5%と低い。欧州では前半が34%、後半が26%と、前半と比較して後半では低下したが、日本と比較して大幅に高い。同年承認の新薬における米国でのブレークスルー指定とオーファン指定は後半に高まっており24)、同時に新興企業が開発企業となる割合も16%から20%へと高まっている。以下ではこれらの特徴と同年承認確率との関係を統計的に分析する。推計モデルは、(3)式の線形確率モデルである。
推計結果は表6の通りである。モデル1からモデル4までは表2と同じ説明変数で、日本または欧州の米国との同年承認確率の要因を分析している。モデル1が最も単純な推計モデルでFDA承認年ダミーのみをコントロールする基本モデルである。モデル2はブレークスルー指定、新薬開発企業の類型などをコントロールするダミー変数を付加し、モデル3は更に15のATC大分類ダミーをコントロールしている。モデル4は、L(抗悪性腫瘍薬、免疫調節剤)及びJ(全身性の抗感染症薬)の分野に推計を限定したサンプルである。他方で、モデル5と6は、日欧を分けて推計している。
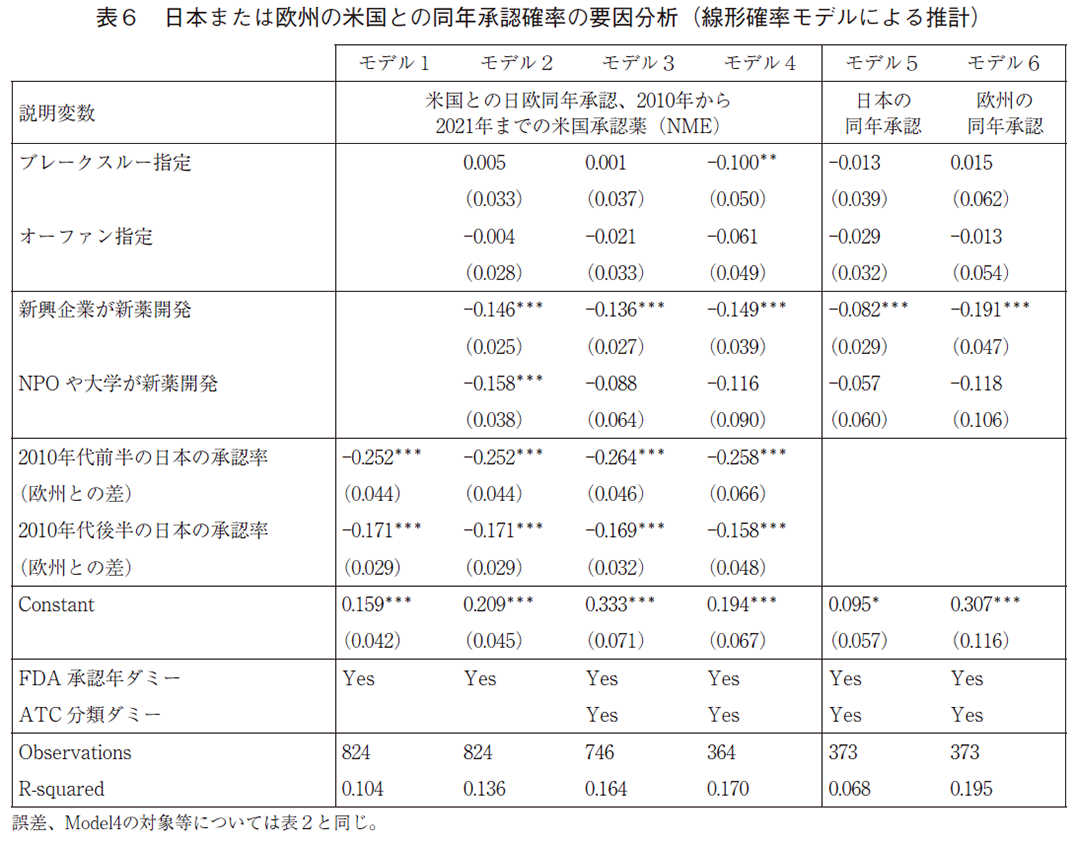
表6のモデル1の推計結果は表5の記述統計を確認しており、日本の米国との同年承認率は欧州の米国との同年承認確率と比較して、前半では25%ポイント低く、また後半では17%ポイント低い。このように日本と欧州の差は低下しているが、表5で確認できるように、その原因は欧州の同年承認率が減少したからであり、日本の同年承認が高まったからではない。なお、COX比例ハザードモデルと異なって前期、後期を直接比較できる。
次に、モデル2によれば、新薬が米国でブレークスルー指定されているかどうか、またオーファン指定をされているかどうかは、日欧とも平均して米国との同年承認の確率と統計的に有意な関係は無い。モデル5と6が示すように、日欧個別の推計モデルからの結果でも同様である。L(抗悪性腫瘍薬、免疫調節剤)及びJ(全身性の抗感染症薬)の分野に推計を限定したモデル4では、ブレークスルー指定は負で有意な係数を持っている。結果は省略しているが、知的財産残存保護期間も有意ではなく、同年承認についてはインセンティブよりも、制度的な障壁の方が重要である可能性を示唆している。例えば、新規性の高いブレークスルーセラピーの場合、米国の迅速承認(Accelerated Approval)制度の対象となっている割合が一定数存在(2010-2021年のFDA承認新薬481品目のうち、ブレークスルー指定は109品目あり、そのうちの41品目が迅速承認されている。)し、日欧との同年承認が生じにくい可能性がある。
ATC分類で領域をコントロールしたモデル3によると、臨床試験の開発企業が新興企業の場合は、同年承認の可能性は約14%ポイント、有意に低くなる。この結果は表2の米国先行承認に限定した承認確率の推計結果と整合的であり、新興企業への資金制約等が国際共同治験の実施も困難にしていることを示唆している。モデル5と6が示すように、米国先行の場合の結果とは異なって、新興企業の負の係数は欧州での承認でより大きい(欧州では19%ポイント減少、日本では9.6%ポイントの減少)。日本では同年承認がもともと低く、同年承認については開発企業が新興企業であることがバインディングな制約となる程度が低い可能性があるが、なお今後の分析が必要である。
9. 結論
本稿では、米国で先行して承認された新薬の、日欧への普及が、新薬の革新性、新薬開発企業の特性、知的財産保護期間などによってどのように影響を受けるか、また日本と欧州の差の有意性と要因を定量的に分析した。更に、米国との同年承認についても同様の分析を行った。本研究では普及年を規制当局の承認年で識別している。主な知見は以下の通りである。
第一に、米国で先行承認された新薬が日欧で承認される確率には、疾患分野の差が大きいが、これをコントロールしても、新薬の革新性(FDAのブレークスルー指定で評価)が高い場合に、日欧で承認される確率は上昇する傾向にある。また、日本の承認確率は欧州と比較して有意に低いが、革新性が高い新薬とそうでない新薬で有意な差は無い。
第二に、国際共同治験等によって、米国との同年承認が欧州では高い水準となっているが、日本の水準は2010年代後半で欧州の3分の1程度と低い。新薬の革新性が高い場合に、日欧での同年承認の確率が高くなる傾向はなく、低下する場合もある。同年承認についてはインセンティブよりも、制度的要因の方が重要である可能性を示唆している。例えば、新規性の高いブレークスルーセラピーの場合、米国の迅速承認(Accelerated Approval)制度の対象となっている場合も少なくなく、日欧との同年承認が生じにくい可能性がある。
第三に、新薬開発企業が新興企業である場合に、日欧における当該新薬の承認確率は大きく低下する。米国先行承認の場合に、日欧平均で、承認確率が約37%低下し、同年承認の確率も約14%ポイント低下する。また、その影響は日本の承認で大幅に大きく、日欧の承認率格差の非常に重要な要因にもなっている。
第四に、米国承認時の知的財産残存保護期間が長い場合に欧州では有意に承認確率が高まる。残存保護期間が1年長い場合に約5%承認確率が高まる。他方で、日本で知的財産保護期間の効果は有意ではない。その原因として、市場の広さや薬価水準、更に薬事規制など知財保護を補完する環境や制度に制約があることが示唆される。なお、日欧いずれにおいても、データ保護期間が重要な場合には、米国承認時の知的財産残存保護期間は各国での独占実施期間を予測しないので、推計結果は過小評価の可能性がある。
このように、日本の新薬承認率は、先行承認国(米国)からの新薬普及においても、国際的な同年承認においても、欧州と比較して有意に低い。日本における基本的な治験環境(低い症例集積性やCRO・SMOの高額費用等)に加え、日本における臨床開発投資へのインセンティブと国際共同治験を含む治験の規制等のあり方に改善すべき点があることを示唆している。その一つとして、開発企業が新興企業である場合に、企業間連携による日本における臨床試験力の強化が重要な課題だと考えられる。
なお、各国の規制当局による承認がなされても市場での上市がなされなければ患者に新薬は届かず、企業も投資の回収を行うことはできないので、承認された新薬の実際の上市の有無とタイミングが重要である。今回の研究において、そのデータの構築も試みたが、利用したデータベースのカバリッジが必ずしも完全でなく25)、国際比較可能性が明確でないことが判明したために、今後の研究課題としたい。
-
1)本稿の研究には医薬産業政策研究所の研究員各位から有益なコメントを頂いたことに感謝申し上げたい。本研究は、科研費基盤B(「創薬イノベーションとインセンティブの研究」、18H00854)の支援も受けて実施した。
-
2)Cockburn, I.M., Lanjouw, J.O., Schankerman, M. 2016. Patents and the global diffusion of new drugs. American Economic Review 106 (1), 136-164.
-
3)医薬産業政策研究所「ドラッグ・ラグ:国内未承認薬の状況とその特徴」政策研ニュースNo.63(2021年7月)、「ドラッグ・ラグ:未承認薬は日本のアンメット・メディカル・ニーズに応えうるか?」、「ドラッグ・ラグ:なぜ、未承認薬が増えているのか?」政策研ニュースNo.66(2022年7月)
-
4)医薬産業政策研究所「ドラッグ・ラグ:日本と欧州の未承認薬状況の比較 -2010~2021年の米国承認薬をもとに-」政策研ニュースNo.67(2022年11月)
-
5)以下では簡略に、それぞれ、「ブレークスルー指定」、「オーファン指定」と呼称する。
-
6)分析では、特許制度と価格規制が内生的に決定されることに対して、国の固定効果をモデルに含めるとともに、操作変数法を用いた分析も行っている。操作変数を用いた分析では、価格規制と特許保護の影響はより強く発現し、外生的に扱われていた従来の分析はそれらの政策制度の影響を過小評価していたことを指摘している。
-
7)Gaessler, F., Wagner, S. 2022. Patents, data exclusivity, and the development of new drugs. Review of Economics and Statistics 104 (3), 571-586.
-
8)今井優也・成川衛. 2022.「日本における新薬開発遅延の背景に関する研究」、RSMP(レギュラトリーサイエンス学会誌)、vol.12 (3), 235-245, Sep 2022.
-
9)新有効成分かどうかはFDAのCenter for Drug Evaluation Research(CDER)の分類によっている。配合剤の場合も新有効成分がある場合のみに新有効成分として認識されている。同じ成分で複数の承認がある少数のケースについては、最初の承認のみを計量分析の対象としている。
-
10)医薬産業政策研究所リサーチペーパーNo.74(2019年10月)「日米欧における薬価の構造とダイナミクス:革新性の反映」、政策研ニュースNo.62(2021年3月)「新薬の比較薬に対する価格プレミアム:日米欧のマッチド・サンプルによる分析」、政策研ニュースNo.64(2021年11月)「新薬の革新性と価格プレミアム-日米独のマッチド・サンプルによる分析-」を参照。
-
11)「世界売上高上位医薬品の創出企業の国籍」、政策研ニュースNo.64(2021年11月)、「世界売上高上位医薬品の創出企業国籍調査を振り返る -品目数の動的推移や創薬の担い手の観点から-」、政策研ニュースNo.64(2021年11月)
-
12)2010年-2018年の330承認の品目、内312承認、96%についてIQVIAのMoleculesとマッチングできた。
-
13)上市をして、医薬品を患者に届けるには、薬としての物質の品質・安定性や製造・製造方法、その設備投資でも先行投資をする必要があり、臨床試験投資とこれらの投資を合わせて臨床開発投資と4.1節では記載する。
-
14)待つことが有利になるのは、不確実性のある臨床開発投資の規模と比較して期待利益が小さい、その不確実性が大きい場合である。
-
15)日欧において先行して承認されるケースもあるが、表1が示すように、その比率は小さく、かつ革新性が高い新薬の比率は低く、本稿の推定では対象にしていない。
-
16)承認が解析時点で未承認であっても今後に承認される打ち切りデータも含めて推計できる
-
17)誤解が生じない箇所では、「ハザードレートの対数」を省略して「ハザードレート」として表記している。
-
18)なお、2010年代の前半と後半で係数値の差は大きいが、その統計的な有意性は10%に届かない。
-
19)ATC分類は、WHOの以下webサイトに基づく。また、ATC分類が付与されていない品目については類縁医薬品から予想されるものを採用した。
WHO Collaborating Centre for Drug Statistics Methodology. ATC/DDD Index 2022 -
20)新興企業は承認取得年が設立年から30年以内、且つ、承認取得前年の売上が5億米ドル未満の企業である(「ドラッグ・ラグ:日本と欧州の未承認薬状況の比較 -2010~2021年の米国承認薬をもとに-」政策研ニュースNo.67(2022年11月))。
-
21)残存保護期間は平均値で12年、下位10%で7年であり、それぞれ1.2年、0.7年延びるとモデル(6)では、5%承認確率が高まる。
-
22)臨床試験の長さに対応した特許保護期間延長の制度も日米欧各国に存在している。但し、延長の対象や期間に差は存在する。
-
23)米国における知的財産残存保護期間を説明変数とする単回帰分析で、日本における知的財産残存保護期間について係数は0.33、定数項が7.1年として推計され、係数は1%有意である。独についても似た結果である(係数0.36、定数項7.9年)。
-
24)米国のブレークスルー指定が2012年7月の法律を根拠として整備された制度であることも2010年代後半に拡大している要因である。
-
25)日本では検査薬(イメージング剤など)は薬価収載されない等である。医薬産業政策研究所主任研究員 吉田晃子氏のご指摘に感謝したい。