目で見る製薬産業 抗悪性腫瘍剤と神経系用剤におけるFDA承認動向の変化-日本の未承認薬増加の背景-
医薬産業政策研究所 統括研究員 飯田真一郎
医薬産業政策研究所 元主任研究員 澁口 朋之
医薬産業政策研究所 主任研究員 吉田 昌生
1. はじめに
抗悪性腫瘍剤や神経系用剤では2010年代後半の日本未承認薬が増加していることが政策研ニュースNo.63(2021年7月)1)や国立がん研究センターの調査2)にて明らかになっており、その実態として日本法人や国内管理人を持たない新興バイオ医薬品企業の製品が多く、国内では開発に着手されていないことが示唆されている。
本稿では、2010年代後半の未承認薬増加の背景について追究するため、2011年~2020年の抗悪性腫瘍剤および神経系用剤のFDA承認薬(新規有効成分)を取り上げ、FDA申請企業やピボタル試験の動向を調査・分析した。
2. 研究方法
国内未承認薬は政策研ニュースNo.63(2021年7月)1)と同様の方法で特定した。本稿ではFDAの承認薬(2011年~2020年)のうち、未承認薬が多い疾患領域3)の抗悪性腫瘍剤(104品目)および神経系用剤(32品目)を分析対象とした。FDA承認薬(新規有効成分、New Molecular Entity; NME)の集計は、Center for Drug Evaluating Research(CDER)による「New Molecular Entity(NME)Drug & Original BLA Calendar Year Approvals」に掲載されている医薬品(診断用薬は除く)を対象とした。薬効分類はWHOのwebサイトを参照4)し、各品目のThe Anatomical Therapeutic Chemical codeをもとに行った。2010年代の前半(2011-2015年、以下、前期)および後半(2016-2020年、以下、後期)に分けて種々比較した。
FDA承認時のピボタル試験はPrescribing Informationの臨床項から特定し、試験参加国等の臨床試験情報はClinicalTrials.govから取得した5、6)。
企業分類はEvaluate Pharmaにて企業設立年を調査し、新興企業は1990年以降に設立した企業とした。1990年以前でも売上1億ドル以下の1社も新興企業に分類した。新興企業以外を製薬企業とした。新興企業の国内開発動向は明日の新薬にて確認した。
3. 結果
抗悪性腫瘍剤のFDA承認動向
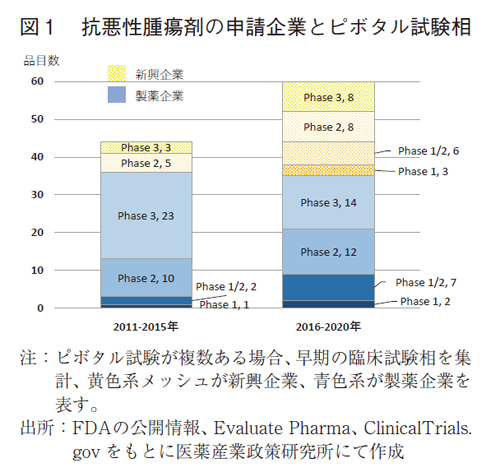
2010年代後半においてはFDAでの全承認薬数や申請企業の変化等が報告されている1、7)。抗悪性腫瘍剤の承認品目の動向を確認するため、前期(2011-2015年)から後期(2016-2020年)への変化を調査した(表1)。特にFDA承認の根拠となる、有効性・安全性を評価する主要な臨床試験:ピボタル試験を分析した。
抗悪性腫瘍剤の申請企業とピボタル試験の臨床試験相の内訳を図1に示す。前期では承認総数44品目のうち、製薬企業が32品目(73%)、8品目(18%)が新興企業であった。後期においては、製薬企業が35品目(58%)、新興企業が25品目(42%)と、製薬企業の品目数が微増の中、新興企業の品目数は3倍以上に増加し、後期品目の4割を占めるに至っていた。抗悪性腫瘍剤においても新興企業が大きく貢献し、承認数の増加に寄与していることが確認された。
ピボタル試験の内訳をみると、前期ではPhase3が最も多く26品目(59%)であった。Phase1/2やPhase1もあったがその品目数は少なく、計3品目(7%)であった。後期ではPhase3は22品目(37%)と比率が低下した。Phase1/2やPhase1が18品目に大きく伸ばし、比率は30%に及んだ。
新興企業ではPhase1/2やPhase1の比率(36%)が高く、Phase2以下では5品目から17品目(68%)に品目数を大きく伸ばしていた。
製薬企業においても総数は横ばいのところ、Phase2以下では13品目から21品目(60%)に増加しており、ピボタル試験の早期化が観察された。
抗悪性腫瘍剤のFDA承認品目においては、新興企業が台頭している特徴並びにピボタル試験の臨床試験相が早期化している特徴が明らかとなった。
ピボタル試験の早期相化の特徴として、患者登録数について前期と後期で比較した(図2)。
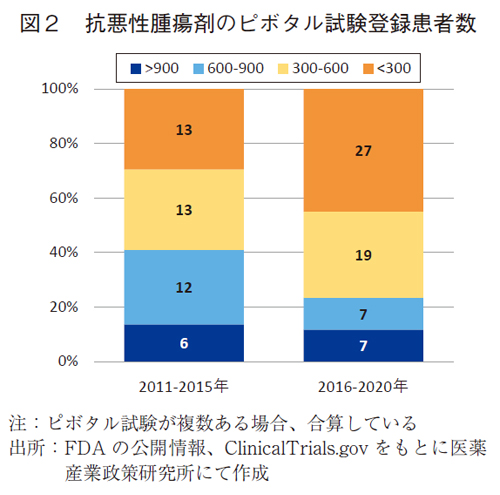
前期の登録患者数(中央値)は472.0例であったが、後期では330.5例と3割程度の縮小が観察された。300例以下の品目数は13品目(29.5%)から27品目(45.0%)と品目数は倍増し、後期の約1/2に迫るまで比率も増加した。抗悪性腫瘍剤のFDA承認品目では、小型化したピボタル試験が増加していることが示された。
ピボタル試験の早期化、小型化の背景を探るため、承認薬の個別化医療の品目を確認した。
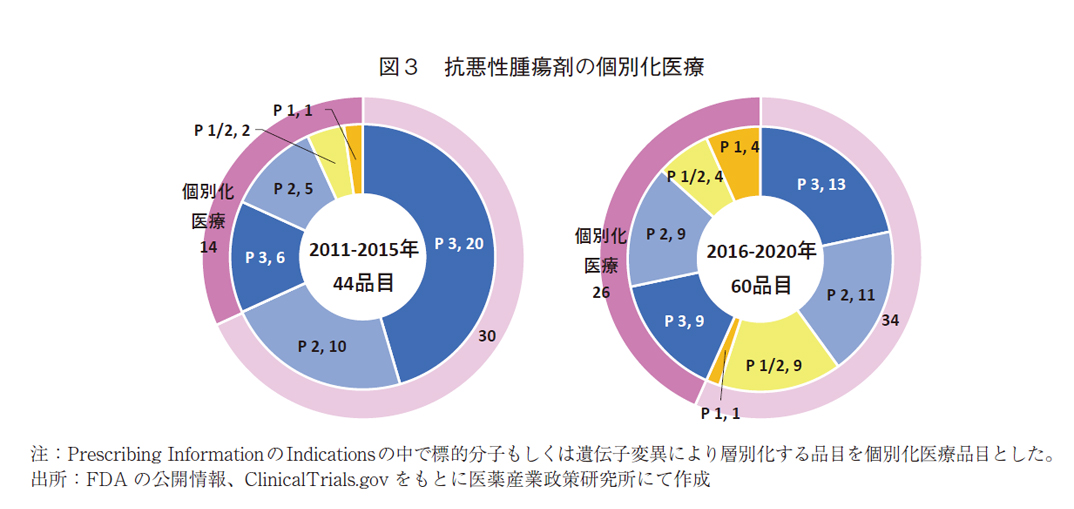
がんのゲノム解析が進展し、がんの原因となる遺伝子変異の解明が進み、分子標的薬が多く開発されている。ドライバー変異遺伝子を診断した上で、その標的に特異的な薬剤による治療(いわゆる個別化医療)が貢献するようになってきている。個別化医療の品目について前期・後期での変化をみると(図3)、前期では14品目(32%)であったが、後期では26品目(43%)と品目数は2倍弱になり、比率も高まっていた。
ピボタル試験の早期臨床試験相(Phase1/2やPhase1)との関係をみたが、個別化医療品目で特に品目数が多い/割合が高いわけではなかった。
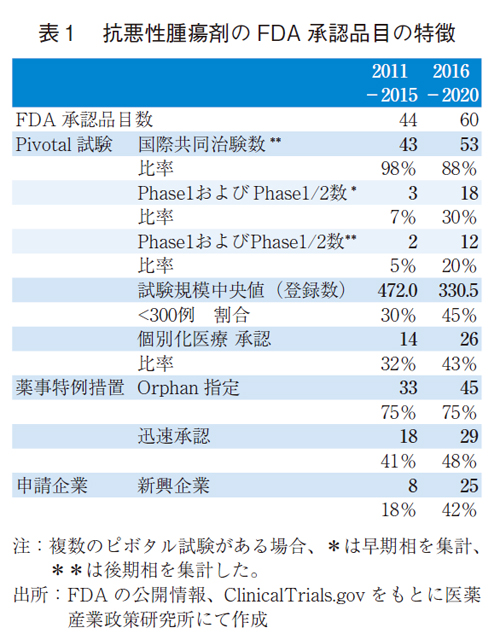
希少がんや希少セグメントを対象とした抗悪性腫瘍剤の開発の増加が想定されることから、Orphan Drug指定品目をみると、比率の変化は見られなかったが、前期では33品目(75%)、後期では45品目(75%)と品目数は増加した(表1)。抗悪性腫瘍剤では、新興企業が台頭し、承認品目ではPhase2より早期臨床試験相での品目数や割合が上がっていることが観察された。その背景には、科学の進展に伴い、小規模でも高い有効性が検証されていることが推察された。
神経系用剤のFDA承認動向
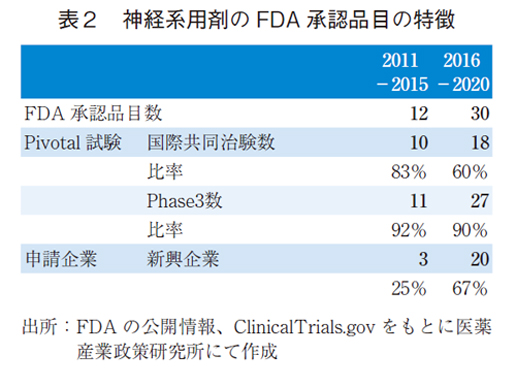
神経系用剤のFDA承認薬についても同様に2011-2015年(前期)および2016-2020年(後期)にて分類し、その特徴を比較した(表2、図4)。神経系用剤では前期で12品目、後期で30品目と後期で増加した。製薬企業では9品目から10品目と横ばいであったものの、新興企業では3品目から20品目に顕著に増加していた。全承認品目の増加は新興企業による品目増加によるものであった。
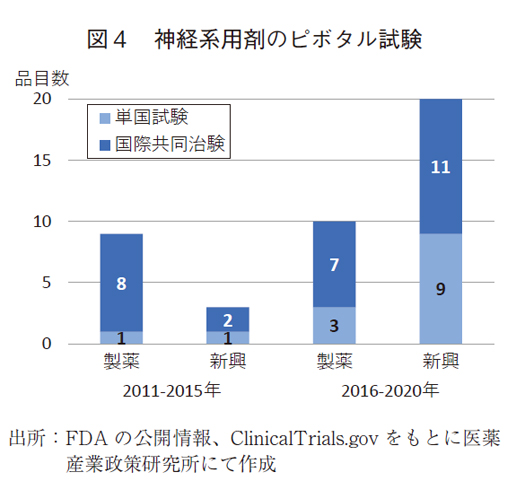
また、ピボタル試験の国際共同治験数にも変化が見られ、前期で10品目(83%)から後期で18品目(60%)であった。国際共同治験の品目数の増加が見られる一方、単国試験が増加したため、国際共同治験の比率は低下していた。なお、神経系用剤では、Phase1/2をピボタル試験とした品目は一つもなかった。
4. まとめと考察
日本未承認薬の要因を追求する上で、海外の承認トレンドとしてFDA承認薬について確認した。
抗悪性腫瘍剤においては、後期(2016-2020年)で品目数の増加がみられた。製薬企業の品目数は横ばいであったが、新興企業による品目数が顕著に増加していた。
ピボタル試験については、早期相のピボタル試験の増加ならびに試験規模の小型化、個別化医療品目の増加が観察された。
がんの薬物治療においては、がんゲノム解析が進み、がんの原因となる遺伝子変異の解明が進んだと共に、これに対する治療薬が数多く開発されてきた。その実用化は2010年代に本格化されていることが確認された。個々の遺伝子変異に応じた標的分子薬による介入は、小さい試験規模であっても有用性が検証されているものと考えられる。個別化医療においては、ヘテロながん集団から、遺伝子変異等の診断により、科学的に均一な集団に対して、標的分子薬の介入を行う。そのため、高い有用性を観察できる確率は高まると共に、小規模かつ早期の臨床試験相での検証が可能となっているものと考えられる。
個別化医療品目以外においても、そのほとんどの品目は、対象とするがんの原因や特徴となる分子を標的としている、いわゆる分子標的薬であるため、個別化医療品目と同様の現象が見られているものと思われた。承認品目のピボタル試験の特性やその担い手の変化の背景には、科学技術の進展が大きく寄与していたものと推察される。
近年、新興企業の活躍が目覚ましい要因については分析に至っていないが、がん領域においては早期臨床相での検証が可能となっていることから、資金調達の負担も下がっていることも後押ししているのであろう。
抗悪性腫瘍剤および神経系用剤ともに、新興企業による品目が後期で顕著に増加していた。新興企業では研究開発資金や資源は潤沢ではなく7、8)、また医薬品開発の全機能を備えているとは考えにくい。その中で新興企業の品目が増加を可能にしている背景の調査には至っていないが、幾つかの環境変化が推察される。いわゆる米国の投資家によるヘルスケアセクターへの投資規模増大により新興企業が研究開発資金を確保できているのであろう。また医薬品開発においては、臨床試験の運営・治験薬や製品の製造などを担うアウトソーシングの充実も新興企業の活躍の場を広げていると考えられる。抗悪性腫瘍剤の個別化医療に見られるように、対象疾患・セグメントが細分化し、基礎研究・臨床開発、そして商業化のバリューチェインを通して、専門性に特化した小さい規模で完結できる領域、いわゆるスペシャリティ領域に変化していることも背景にあることも想定される。変化の背景については、更なる分析が必要であろう。
日本の未承認薬を減らすためには、製薬企業による国際共同治験の推進のみならず、新興企業の品目に対する手当の必要性が示唆された。日本への最新医薬品のアクセスを維持・確保するためにも、海外動向の変化を捉えた政策に進化することを期待したい。
-
1)医薬産業政策研究所「ドラッグ・ラグ:国内未承認薬の状況とその特徴」政策研ニュースNo.63(2021年7月)
-
2)健康・医療戦略推進本部「第1回 医薬品開発協議会 資料2-6」(2020年10月27日)
-
3)医薬産業政策研究所「ドラッグ・ラグ:未承認薬は日本のアンメット・メディカル・ニーズに応えうるか?」政策研ニュースNo.66(2022年7月)
-
4)
-
5)医薬産業政策研究所.「近年の国際共同治験の参加国の分析-臨床試験登録システムClinicalTrials.govを基に-」政策研ニュース No.58(2019年11月)
-
6)医薬産業政策研究所「近年における国際共同治験の動向調査」政策研ニュースNo.66(2022年7月)
-
7)IQVIA “Global Trends in R&D: Overview through 2020” (May 2021)
-
8)医薬産業政策研究所「ドラッグ・ラグ:なぜ、未承認薬が増えているのか?」政策研ニュースNo.66(2022年7月)