Topics ドラッグ・ラグ:なぜ、未承認薬が増えているのか?
医薬産業政策研究所 統括研究員 飯田真一郎
医薬産業政策研究所 元主任研究員 澁口 朋之
医薬産業政策研究所 主任研究員 吉田 昌生
要約
- 海外での新薬創出において新興企業が活躍している中、抗悪性腫瘍剤の日本未承認薬数の増加は、新興企業品目の増加、ピボタル試験の早期臨床相化、そしてこれら品目の早期臨床試験への低い日本地域組入れ率に起因していた。
- 神経系用剤の未承認薬増加においても、新興企業の未承認品目が増加し、ピボタル試験への低い日本地域組入れ率が要因であった。
- 新興企業は、収益がない中でピボタル試験に日本地域が組み入れられず、日本市場展開の優先度は低いことが示唆された。
- 本邦への医薬品アクセスを確保するためには、海外新興企業の医薬品について、日本での医薬品開発および市場展開を促進する政策が期待される。
1. はじめに
「未承認薬の増加」は本邦の最新医薬へのアクセスにとって重大な課題であり、解消に向けた取組みが必要とされる。ここ最近、2010年代後半に未承認薬数及び未承認薬比率が増加していることが政策研ニュースNo.63(2021年7月)1)や国立がん研究センターの調査2)にて明らかになっており、その実態として日本法人や国内管理人を持たない新興バイオ医薬品企業の製品が多く、国内では開発に着手されていないことが示唆されている。前報3)においては、これら未承認薬は臨床的に重要度の高い医薬品が多くを占めることが確認され、未承認薬の増加は日本の最新医薬品アクセスにとって見過ごせない課題としてその原因と対策の検討が必要とされる。
IQVIA4)によると、FDAでNovel Active Substancesとして承認された品目の内、新興バイオ医薬品企業により上市された品目が年々増加し、2020年には4割に達したことが報告されている。背景となるFDAの新薬承認状況は次報5)も併せて参照頂きたいが、医薬品開発における担い手の変化も未承認薬の増加に影響していることも考えられる。
本稿では、2010年代後半の未承認薬増加の背景と共にその要因について追究するため、2011年~2020年の抗悪性腫瘍剤および神経系用剤のFDA承認薬(新規有効成分)を取り上げ、未承認薬の特徴を調査・分析すると共に解消に向けた方向性を考察したい。
2. 研究方法
国内未承認薬は政策研ニュースNo.63(2021年7月)1)と同様の方法で同定した。本稿ではFDAの承認薬(2011年~2020年)のうち、未承認薬が多い疾患領域3)の抗悪性腫瘍剤(104品目)および神経系用剤(32品目)を分析対象とした。FDA承認薬(新規有効成分、New Molecular Entity ;NME)の集計は、Center for Drug Evaluating Research(CDER)による「New Molecular Entity(NME)Drug & Original BLA Calendar Year Approvals」に掲載されている医薬品(診断用薬は除く)を対象とした。薬効分類はWHOのwebサイト6)を参照し、各品目のThe Anatomical Therapeutic Chemical codeをもとに行った。2010年代の前半(2011-2015年、以下、前期)および後半(2016-2020年、以下、後期)に分けて種々比較した。それぞれ2015年末時点および2020年末 時点で国内で承認されていない品目を未承認薬とした。
FDA承認時のピボタル試験はPrescribing Informationの臨床項から同定し、試験参加国等の臨床試験情報はClinicalTrials.govから取得した7、8)。
企業分類はEvaluate Pharmaにて企業設立年を調査し、新興企業は1990年以降に設立した企業とした。1990年以前でも売上1億ドル以下の1社も新興企業に分類した。新興企業以外を製薬企業とした。新興企業品目の国内開発動向は明日の新薬にて確認した。
未承認薬増加への影響度は、統計解析ソフトStata/IC 14.0 for Windows(Stata Corp LP, College Station, TX, USA)を使用し、ロジスティック回帰分析により推定し、最小二乗法を用いた線形モデルにより頑健性を確認した。
3. 分析結果
3-1. 抗悪性腫瘍剤の未承認薬
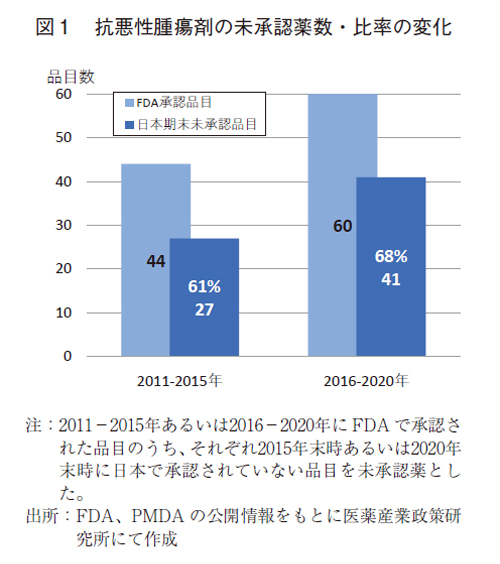
2011-2015年(前期)および2016-2020年(後期)にFDAで承認された抗悪性腫瘍剤(それぞれ44品目および60品目)につき、未承認薬の現状を確認した(図1)。前期および後期の品目のうち、国内未承認薬はそれぞれ27品および41品であった。未承認薬品目数は増加し、未承認薬比率もまた、61%から68%に増加した。欧米の新薬全体での未承認薬増加1)と同様に増加した事が確認された。
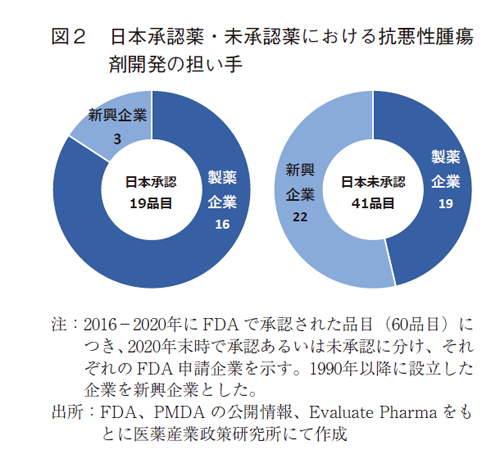
後期の品目について、2020年末時の日本の承認有無で分類し、FDA申請企業の割合を調査した(図2)。日本で承認された19品目では16品目(84%)は製薬企業が担っていた。一方、日本で未承認であった41品目では22品目(54%)は新興企業の品目であることが示された。後期の抗悪性腫瘍剤の未承認薬では新興企業の品目数の比率が高く、未承認薬の増加との関連性が示唆された。
3-2. 抗悪性腫瘍剤の日本未承認薬の内訳
抗悪性腫瘍剤の日本未承認薬の品目数および比率が後期に増加していることを示した。同時期のFDA承認薬では、申請企業やピボタル試験の特徴が変化している5)。未承認薬増加の要因に迫るため、未承認薬品目数の増加におけるFDA申請企業やピボタル試験の関連性を追究した。
-未承認薬では新興企業の品目が顕著に増加している-
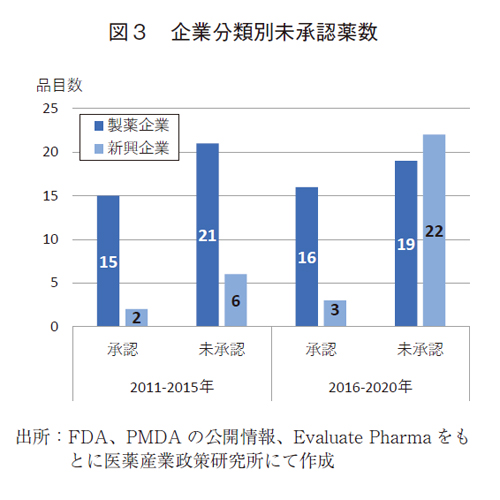
図3において前期から後期における申請企業毎の未承認薬数の変化を示す。製薬企業の品目では、承認薬および未承認薬ともに前期と後期に大きな変化は見られず、日本未承認薬では21品目から19品目と微減であった。新興企業の品目では、承認薬が微増であったが、日本未承認薬については、前期から後期にかけて6品目から22品目に顕著に増加していた。
-ピボタル試験への日本地域組入れは増加しているが、未承認薬では日本地域組入れが少ない-
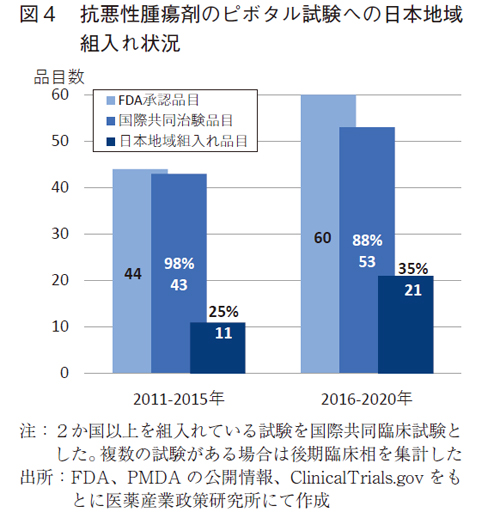
海外に遅れることなく日本で承認されるためには、海外承認時のピボタル試験が国際共同治験であり、その試験に日本地域が組入れしていることが重要である。図4では、ピボタル試験における日本地域の組入れ状況を示す。
抗悪性腫瘍剤のFDA承認薬においては、多くのピボタル試験は国際共同治験となっている。前期では43品目(98%)、後期では53品目(88%)が国際共同治験であった。後期では品目数は増加しているものの、米国での単国試験が増加し、国際共同治験比率が低下していた。
FDA承認品目の国際共同治験比率が低下する中ではあったが、日本地域が組入れられているピボタル試験は、前期では11品目(25%)、後期では21品目(35%)と品目数・組入れ率ともに増加した。
日本への最新薬剤のアクセスを確かなものにするため、「国際共同治験に関する基本的考え方」(2007年、厚生労働省)の発出や国際共同治験への日本地域組入れに各製薬企業の尽力もあり(7、8)、組入れの品目数・比率とも増加していたことから、一定の成果が見られていた。
しかしながら、FDA承認薬のうち、日本組入れがないピボタル試験の品目数は、21品目以外の39品目(65%)と依然として多く、開発ラグや未承認薬となる可能性が危惧された(図4)。
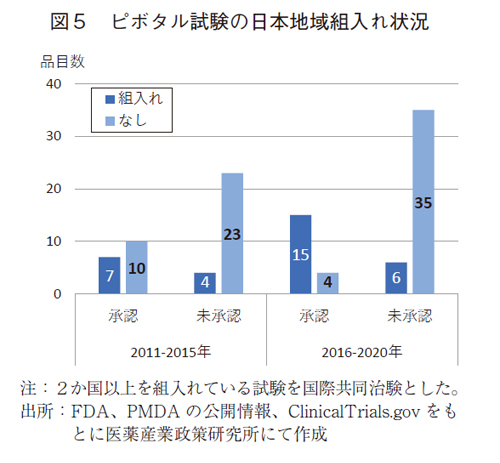
国際共同治験への組入れ率は増加しているにも関わらず、日本未承認薬は増加しているため、抗悪性腫瘍剤の国際共同治験について各国の組入れ状況を確認し、承認薬と未承認薬の相違を分析した(図5、図6)。
日本承認薬について、前期と後期の推移をみると、日本組入れ品目は7品目から15品目に倍増し、組入れなしは10品目から4品目に減少した。承認薬では国際共同治験への日本組入れが進んでいることが明らかであった。
一方、日本未承認薬では、組入れ品目は4品目から6品目に増加しているものの、組入れなしは23品目から35品目に顕著に増加した。後期の未承認薬においては、組入れなしの比率は、85%(35/41品目)にも達して(図10)いた。
後期においては全体の組入れ率が35%であるにも関わらず(図4)、承認率は32%(図1)に留まっていた。組入れは日本承認薬で倍増していたが、承認薬総数は倍増してはいなかったことが承認率が低いことに影響していると考えれらる。また、日本組入れがあったが未承認である6品目をみると、4品目では2021年末迄に承認されていたことから、後期の組入れ品目の一部では承認に遅延があった。その内訳は、新興企業品目で日本の開発者が変わることによる遅延や早期ピボタル試験相のため、日本では追加試験を実施し、遅延が発生している可能性が考えられた。
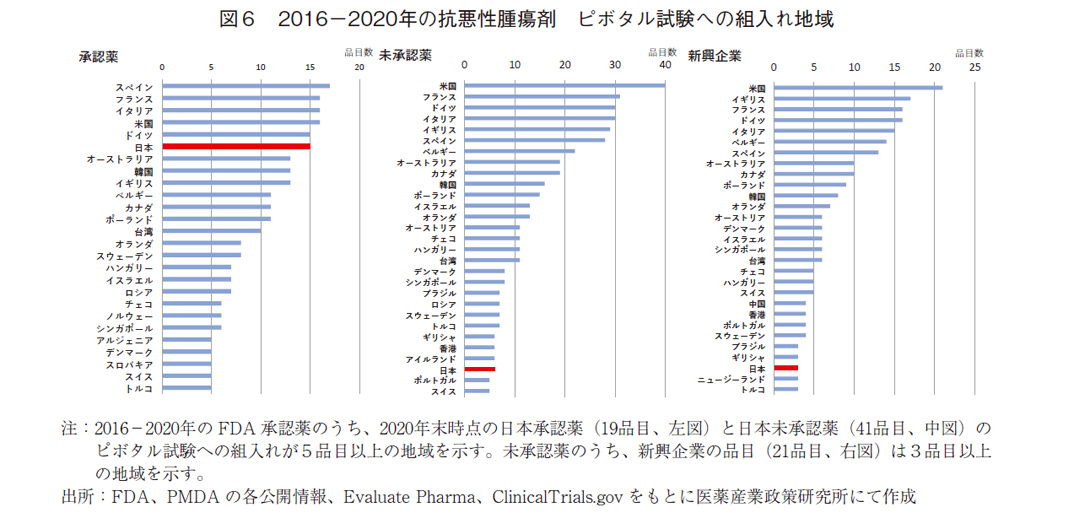
2016-2020年の60品目について、各国の組入れ状況を図6に示す。日本承認薬19品目を見ると18品目が国際共同治験であり、日本の組入れ品目数(15品目)はG7各国と同等であった。なお、韓国(13品目)や台湾(10品目)においても多く組み入れられており、アジア諸国においても国際共同治験が推進されていた。
日本未承認41品目では、34品目が国際共同治験、6品目は単一国試験であった。それらの組入れ国(単一国を含む)を見ると、日本を除くG7各国では30品目程度は組入れられているのに対し、日本は6品目に留まっていた。アジアでは、韓国(16品目)および台湾(11品目)が組入れられており、日本はこれらの地域より明らかに組入れが少なかった。人口が少なく、医薬品市場も小さい韓国・台湾より少ないことから、日本地域の薬事・臨床試験環境にこれらの国と相違があることが示唆された。
新興企業についてみると未承認薬の21品目中、19品目は国際共同治験を実施していた。実施国では、未承認薬全体の傾向と同様であった。米国は全て含まれ、欧州主要国も15品目程度は組入れられていたが、日本はわずか3品目であった。アジアでは韓国(8品目)が最も多く、シンガポール、台湾、中国、香港に次いで日本は6番目の品目数であった。
-未承認薬では、早期ピボタル試験相の品目が増加している-
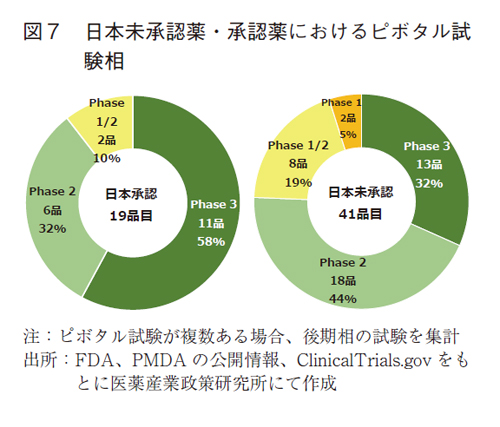
FDA承認品目の後期ではPhase1/2やPhase1がピボタル試験となる品目が増加していた5)。この影響につき、後期の日本未承認薬における各臨床試験相の割合を図7に示す。国内承認された19品目ではFDAのピボタル試験はPhase3が11品目(58%)と中心であった。Phase2は6品目(32%)、Phase1/2も2品目で確認された。国内未承認薬ではPhase1の2品目に加え、Phase1/2が8品目と、早期臨床試験相(Phase1/2、Phase1)の比率は24%と承認薬に比べて高かった。Phase2もまた、18品目(44%)とその比率は高く、Phase3は13品目(32%)と比率は低かった。
未承認薬ではPhase2を含めた早期臨床試験相の品目比率は68%(28/41品目)と承認薬の42%(8/19品目)に比べ、比率が高いことが示された。
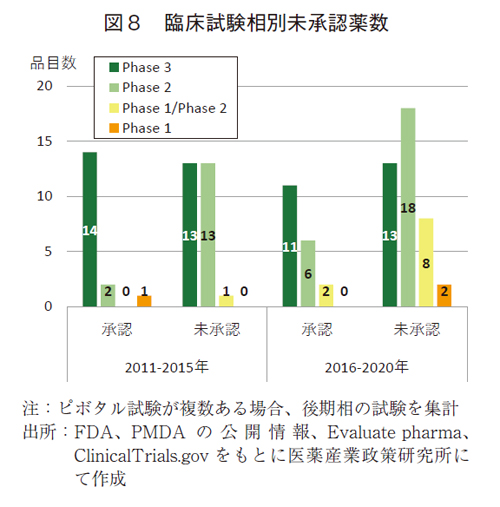
前期から後期の推移をみると(図8)、Phase3での未承認薬は13品目で横ばいのところ、Phase2(13から18品目)やPhase1/2(1から8品目)の未承認薬が増加した。後期ではPhase1の2品目も観察された。承認薬においてもPhase2やPhase1/2がそれぞれ6品目および2品目に増加したが、Phase3品目(11品目)は減少した。
前期から後期への変化を、後期臨床相(Phase3)と早期臨床相(Phase2、Phase1/2、Phase1)に分けて未承認薬をみると、後期相は13品目から変化なく、早期相は14品目から28品目に倍増した。
未承認薬での臨床試験相の変化を新興企業と製薬企業に分けて確認した(図9)。
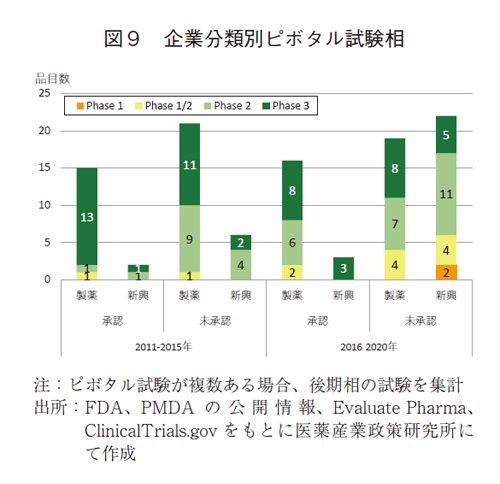
前期と後期の未承認薬について、新興企業の品目を比較すると、6品目から22品目と、16品目の増加が確認された。また後期のピボタル試験相はPhase22が最も多く11品目に増加し、Phase1/2およびPhase1で6品目と早期臨床試験相の増加が見られた。多くの未承認薬は、Phase2より早期の臨床相で計17品目(77%)であった。製薬企業では、承認薬も未承認薬も品目数には大きな変化は見られなかったが、Phase1/2の未承認薬が増加していた。早期臨床相は58%(計11品目)とその比率は増加した。
-新興企業の品目・早期臨床試験相の品目で日本地域の組入れ率が低い-
未承認薬増加の要因として、新興企業・早期臨床相・組入れ率が低いことが明らかになってきたが、これらの関係を探るため、ピボタル試験への組入れ状況を企業分類および臨床相に分けて、組入れ状況を確認した(図10)。
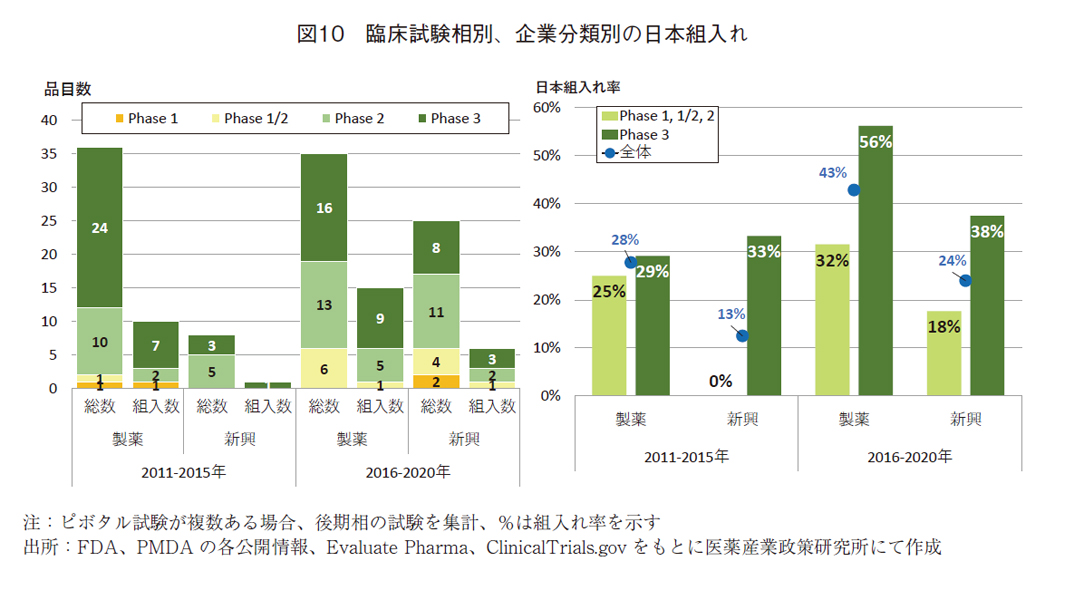
製薬企業の組入れは、前期計10品目(28%)から後期計15品目(43%)と、品目数・組入れ率ともに増加していた。新興企業においても前期1品目(13%)から後期計6品目(24%)に増加したものの、製薬企業の品目に比べて組入れ率は低かった。
臨床試験相別に分解して組入れをみると、製薬企業のPhase3では7品目(29%)から9品目(56%)と後期には品目数・率ともに増加していた。Phase2の組入れにおいても2品目から5品目へ増加していた。その一方、後期におけるPhase1/2への組入れは1品目のみであった。早期臨床相(Phase1、1/2、2)の組入れ率は、25%(前期)から32%(後期)と増加したが、後期臨床相に比べて低かった。
新興企業でのPhase3への組入れは、前期では1品目(33%)であったのに対し、後期では3品目(38%)に増加した。Phase2では2品目、Phase1/2では1品目、Phase1では0品目と少なかった。後期の早期臨床試験相(Phase1、1/2. 2)の組入れ率は、18%と前期に比べて増加したものの、後期臨床相や製薬企業の早期臨床相に比べて低かった。
前期と後期でピボタル試験への日本組入れ率は増加していたものの、新興企業の品目および早期臨床試験相で日本地域の組入れ率が後期で低いことが明らかとなった。
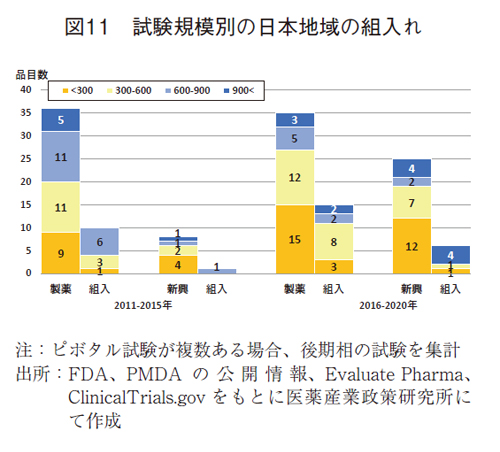
また、FDA承認薬ではピボタル試験規模の縮小が観察されていた5)。日本地域の組入れとの関係をみると、後期で300例以下の試験への組入れ率が、製薬企業の品目で20%(3/15品目)、新興企業の品目で8%(1/11品目)と低いことが示された(図11)。前期においても300例以下の試験への組入れ率は低かった(それぞれ11%と0%)が、後期で小規模試験数が多く、小規模比率が高いことは、日本の組入れ率が低いことに影響を及ぼしていることは想像に難くないであろう。
FDA承認品目では前期から後期に個別化医療の品目数や割合が増加5)したが、日本承認・未承認で個別化医療の品目を比較したところ、大きな差は認められなかった(データ省略)。
-未承認薬増加への影響度は、「日本地域の組入れなし」が大きい-
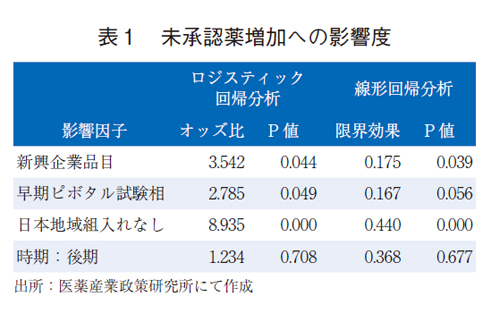
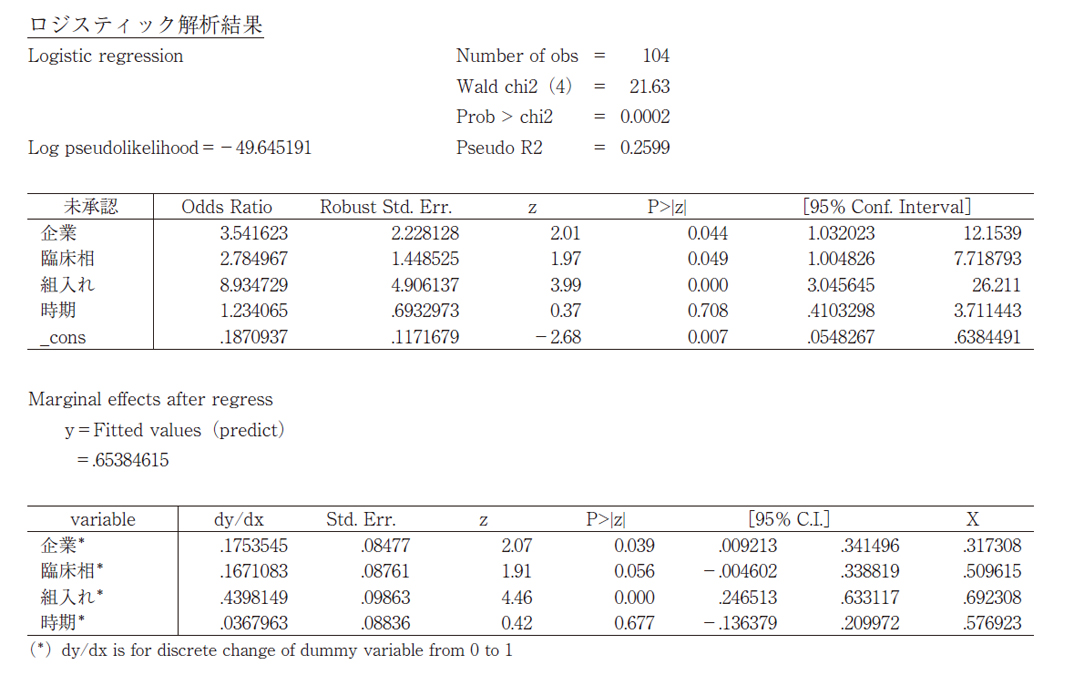
後期における未承認薬の増加の内訳として、1)新興企業品目の増加、2)ピボタル試験相の早期化、3)ピボタル試験相への低い日本組入れ率、が起こっていることが挙げられた。これらの要素につき、未承認薬の増加への影響度を統計手法により推定した(表1)。
104品目のデータを用いたロジスティック回帰分析(付表1)では、「新興企業品目」はP値0.044で有意、「早期ピボタル試験相」はP値0.049で有意、「日本地域組入れなし」はP値0.000で有意、と3つの影響因子において統計学的な有意差が認められた。
それぞれのオッズ比から、日本地域を組入れない場合は組入れに比べて、未承認薬となるオッズ比が8.9倍と推定された。新興企業品目は製薬企業品目に比べて、未承認薬となるオッズ比は3.5倍、ピボタル試験相のPhase2以下の場合は、Phase3に比べて2.8倍のオッズ比であった。オッズ比が1から離れているほど、比較する二群間で顕著な差があると解釈することができるため、ピボタル試験相への日本組入れ率の低下が未承認薬の増加の要因として大きな影響を与えていると考えられた。
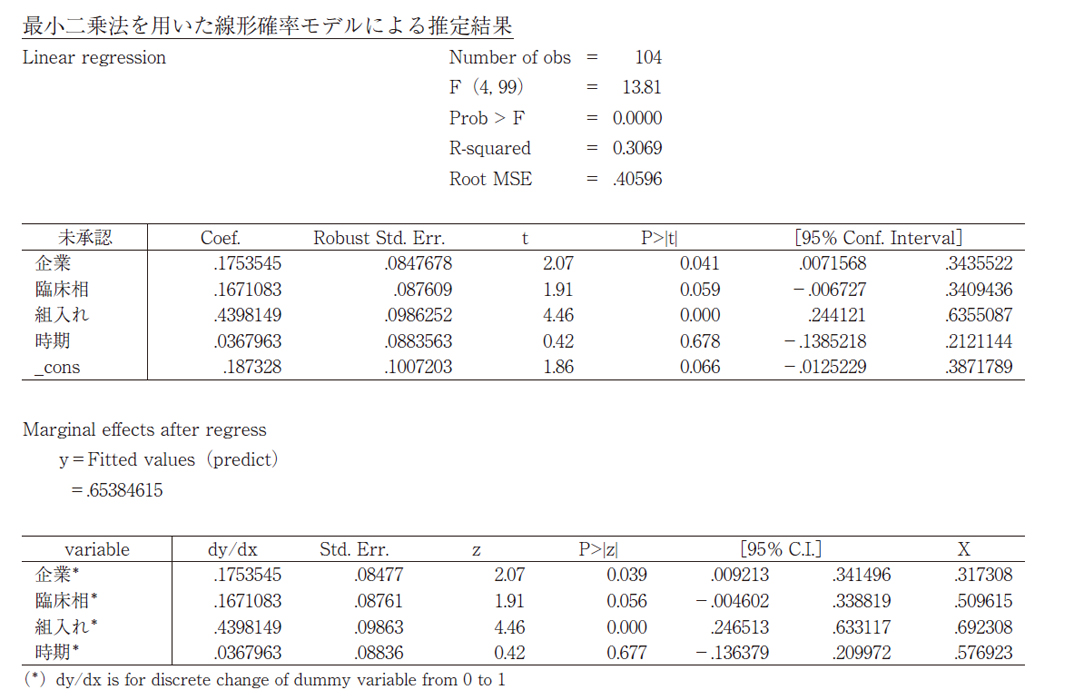
分析の頑健性をチェックするため、最小二乗法を用いた線形確率モデルによる推定も行った。その結果、「新興企業品目」はP値0.039で有意、「早期ピボタル試験相」はP値0.056で有意、「日本地域組入れなし」はP値0.000で有意、といずれも統計学的な有意性が認められた。
それぞれの限界効果を比較すると、日本地域を組入れない場合は組入れる場合に比べて、未承認薬になる確率が44%高いことがわかり、評価した3つの因子の中では未承認薬となる確率への影響度が最も高かった。新興企業品目や早期のピボタル試験相においても、限界効果は18%と17%と推定された。
以上の統計解析結果から、未承認薬増加の要因として、ピボタル試験へ日本組入れがないことの影響が大きく、次いで新興企業、早期臨床試験相であった。
なお、時期(前後期)のような変化するマクロ要因には有意性は検証されなかった。
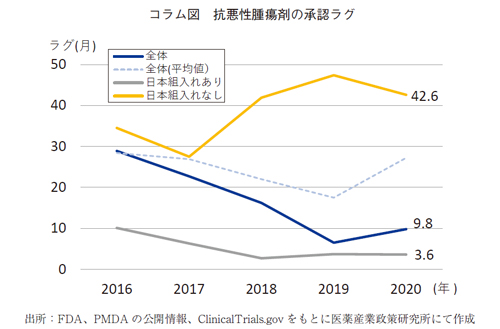
コラム ピボタル試験への日本組入れの効用
ピボタル試験への日本組入れは、承認ラグの視点からも、その重要性は見過ごせない。今回分析対象とした抗悪性腫瘍剤のうち、国内で承認された品目について、ピボタル試験への組入れ有無によるラグを確認した。
抗悪性腫瘍薬の国内承認品目(2016-2020年、それぞれ9、5、8、7、7品目)の承認ラグ(PMDA承認とFDA承認の差)を調査した。各年の品目全体では、2016年の中央値で28.9か月だったものが2020年では9.8か月と短縮している。品目数が少ないため、平均値で確認すると2020年でも27.2か月であった。これをピボタル試験への日本組入れあり・なし(それぞれ中央値)でみると、ラグは明らかに二相性であった。組入れありは2016年の10.1か月からさらに短縮し、2020年には3.6か月であった。一方、組入れなしは2016年で34.5か月のところ、ラグが拡がる傾向にあり、2020年では42.6か月であった。
ピボタル試験に日本が組み入れられないことにより、約3年半の承認ラグが観察され、日本組入れの重要性が示された。
3-3. 神経系用剤の日本未承認薬の内訳
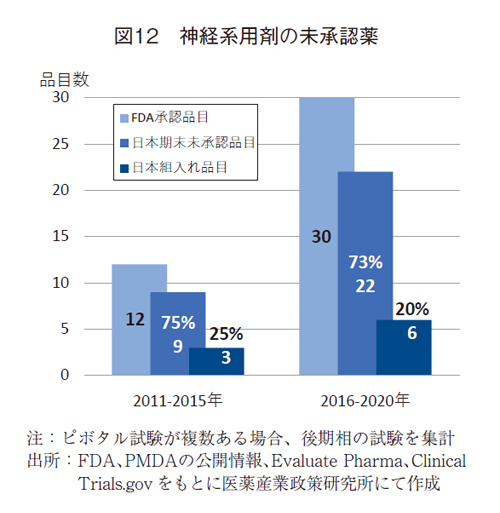
2011-2015年および2016-2020年にFDAで承認された神経系用剤(12品目および30品目)について、その特徴と日本未承認薬の状況を、抗悪性腫瘍剤と同様にまとめた(図12)
-未承認薬品目数が増加、日本地域の組入れ率は低下している-
神経系用剤のFDA承認品目数は12品目から30品目へと2倍以上に増加していた。日本未承認薬は、FDA承認薬数の増加に比例し、9品目(73%)から22品目(75%)に大きく増加した。未承認比率は増加することはなかったが、前期・後期とも高く、7割を超えていた。
ピボタル試験への日本組入れ状況は、前期で3品目(25%)、後期で6品目(20%)と組入れ品目は増えたものの、組入れ比率は低下した。いずれの時期でも低い組入れ率であった。
ピボタル試験の状況をみると、国際共同治験は10品目から18品目に増加したが、後期で単国試験が増加したため、国際共同治験の比率は83%から60%に低下した。なお、ピボタル試験相はPhase1/2以下はなく、Phase3が主体であり、前期・後期ではそれぞれ11品目(92%)と27品目(90%)と9割を超えていた5)。
-未承認薬では、新興企業の品目が増加し、ピボタル試験への日本地域組入れ率が低い-
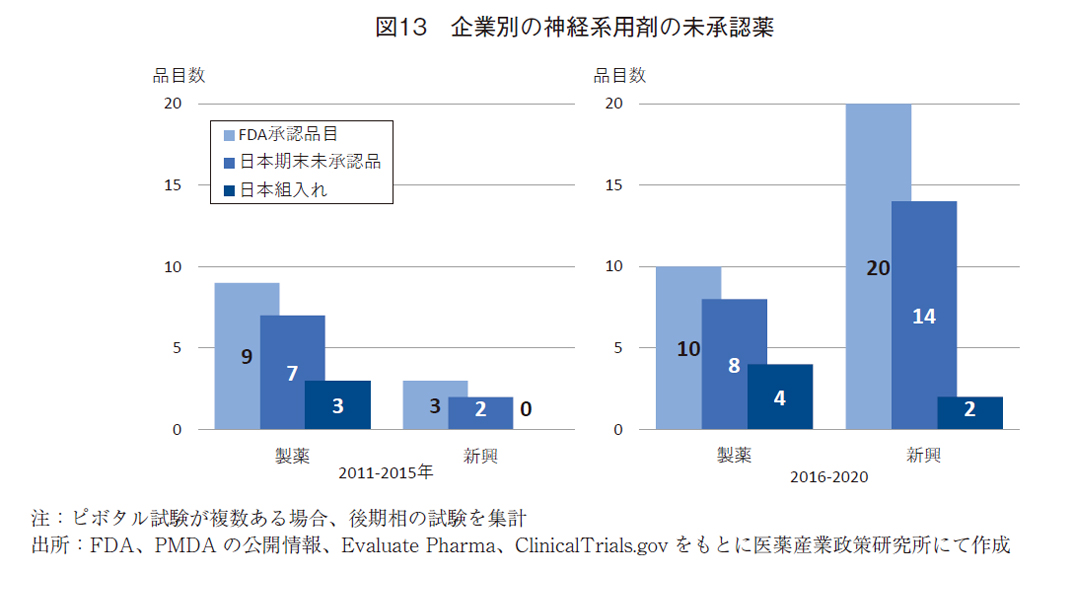
FDA承認薬の企業別状況をみる(図13)と、前期では製薬企業9品目(75%)、新興企業3品目と製薬企業の品目が主体であった。後期では製薬企業10品目(33%)、新興企業20品目(67%)と主体は新興企業の品目に移っていた。すなわち、後期の品目の増加は、抗悪性腫瘍剤と同様に新興企業の品目の増加であることが明らかであった。
前期から後期への日本未承認薬の推移では、製薬企業で7品目(78%)から8品目(80%)に、新興企業で2品目(67%)から14品目(70%)と、未承認比率には大きな変化はなかったが、新興企業の品目が大幅に増加した。
前期から後期へのピボタル試験への日本地域の組入れの推移は、製薬企業で3品目(33%)から4品目(40%)に、新興企業で0品目から2品目(10%)に微増していた。新興企業での未承認薬において、ピボタル試験への日本地域の組入れは10%であったが、十分ではないことは明らかであった。
神経系用剤のFDA承認薬についても、抗悪性腫瘍剤と同様に、後期において新興企業の品目が増加していた。日本未承認薬の比率は前後期で変化なかったが、品目数の増加は確認された。新興企業品目のピボタル試験では単国試験も多く、抗悪性腫瘍剤でみるように、日本地域が組入れられていないことが未承認薬数の増加に大きく影響していると考えられた。
抗悪性腫瘍剤や神経系用剤の未承認薬増加には新興企業の影響が大きい。そこで、新興企業についてはその事業における特徴や事業環境を分析した。
3-4. 新興企業の事業における特徴と環境
2011-2020年に抗悪性腫瘍剤をFDAに申請した新興企業は29社(33品目)であったが、企業情報取得が可能であった26社について、新興企業の設立からFDA承認までの経緯を追った。なお、1社は上場前に買収されていた。
-設立から上市まで15年かかっている-
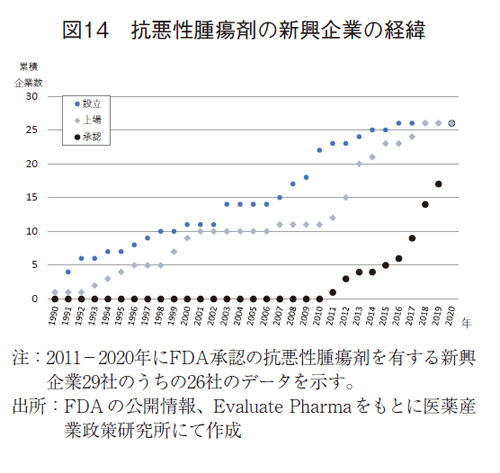
26社では、23社(88%)は米国、他の3社はアイルランド、ドイツ、中国の企業であり、多くは米国拠点の企業であることが確認された。企業設立の中央値は2003/10/31、株式上場の中央値は2012/1/27、FDAの承認年の中央値は2018/12/8であった(図14)。26社個社毎の中央値は、設立から上場まで4年3か月、設立からFDA承認まで14年11か月、上場後からでも6年9か月の時間を要していた。
-上市前の新興企業では売上収益がない-
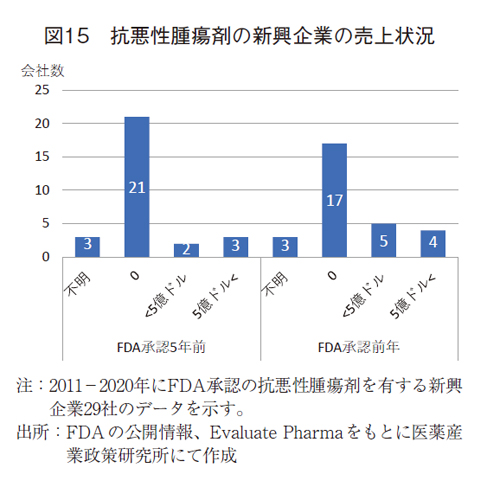
新興企業では商業化に至っていないことが想像されることから、FDA承認の前年と5年前での医薬品売上を確認した。26社では、FDA承認前年で17社(65%)、FDA承認5年前で21社(81%)において、医薬品売上は発生していなかった(図15)。
26社のうち、3社および4社(それぞれ5年前および前年)では5億ドル以上の売上を計上していたが、調査対象新薬に先行して新薬を上市しているケースや製品導入によるものであった。
会社設立から新薬の上市までは約15年の時間を要するとともに、承認前ではほとんどの企業で売上収益がないことが確認された。これらの企業では長期間にわたり研究開発を進めるための資金は潤沢ではなく、外部からの資金調達が必要であることが改めて認識された。
-日本医薬品市場の相対的な魅力度は低い-
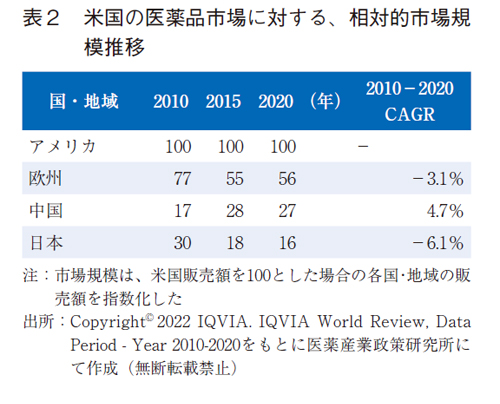
研究開発資金が潤沢にない新興企業にとって研究開発投資の優先度設定は死活問題であろう。新興企業の9割方は米国企業であり、米国での収益最大化は第一優先であることは想像に難くない。その状況下、研究開発の効率性と製品として成功した際の期待収益のバランス、いわゆる投資対効果から、臨床開発の組入れ国の拡大などを視野に入れていることが考えられる。そこで各国の医薬品市場は、米国新興企業からどのように映っているかを類推するため、米国市場に対する相対的な市場規模の推移を描出した(表2)。米国の医薬品市場規模を該当年毎に100とした際に、各国あるいは各地域の医薬品市場規模を指数化した。
欧州、中国、日本で比較すると、中国のみで成長性がみられ、CAGRは4.7%であった。欧州と日本はマイナス成長であり、欧州-3.1%、日本に至っては-6.1%であった。規模の面では欧州は2020年でも米国の半分以上(56)はあったが、日本に至っては16と米国の7分の1強程度であった。
新興企業にとって足場である米国の市場規模と成長性を基準とすると、日本の市場は規模も小さく、マイナス成長に映ることが示された。この10年の推移をみると、日本の医薬品市場は魅力的なマーケットとは捉えがたく、新興企業にとって投資優先度は低い可能性が推察された。
図13にて新興企業のピボタル試験への日本地域の組入れ率が低く、特にPhase2以下の早期臨床試験で低いことが示されている。Phase2がピボタル試験となりえる抗悪性腫瘍剤の領域においては、Phase2試験への日本組入れの投資優先度が低いことは新興企業にとって合理的である可能性はあるものの、日本での最新医薬品のアクセスにおいては大きな課題となることが懸念される。
3-5. 新興企業品目の日本での開発動向
2011-2020年にFDAで承認された抗悪性腫瘍剤および神経系用剤のうち、新興企業品目はそれぞれ33品目(29社)および23品目(21社)であった。これらの新興企業品目について、最新の国内開発状況(2022年4月末時点)を調査した。
新興企業品目は、FDA承認取得の前後を境に、大きく3通りのビジネス形態がとられていた。一つ目は製薬企業によるM&A、二つ目は日本テリトリーの製品導出、三つ目は自社による日本市場開拓等であった。これらに分類し、薬剤の開発状況をまとめる(図16)
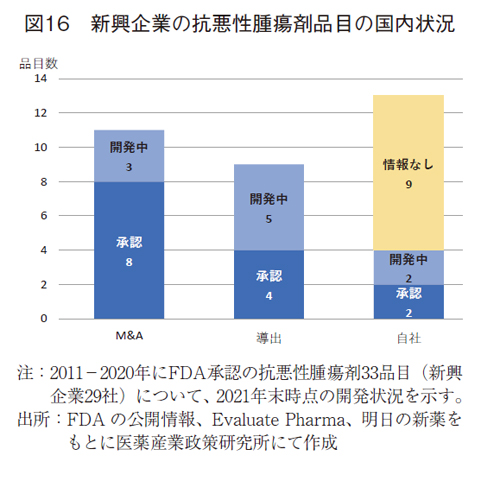
抗悪性腫瘍剤の33品目(29社)について動向を分類した。M&Aの11品目(9社)については、日本企業1社を含む、いわゆるグローバル製薬企業によりFDA承認の1-2年後に企業買収されていた。これらの品目については、国内で承認(9品目)あるいは開発中(3品目)であった。
導出の9品目はグローバルあるいは日本テリトリーにおいて導出されていた。導出時期は同様にFDA承認の1-2年後が多かった。日本テリトリーの導出先の多くは日本企業であった。これらの品目も承認(4品目)あるいは開発中(5品目)であり、M&Aに比べると承認の割合が低かった。
自社の13品目では、複数製品を持つ新興企業では、日本法人を設立する企業も観られ、承認2品目、開発中2品目であった。しかし多くの品目については、情報なし(9品目)と、2022年4月末時点で開発されている様子は確認できなかった。なお、情報なしは調査したデータベースに記載がない、国内臨床試験を実施した経緯はあるが続報なしあるいは中止の品目を含んでおり、医薬品の科学的な有用性が検証されなかったものも含まれている可能性も推察された。
新興企業の品目では、M&Aや導出といった事業母体が変わるプロセスも入るため、日本への事業展開においては、企業-企業間の交渉期間もまた承認ラグとなりうることが示唆された。
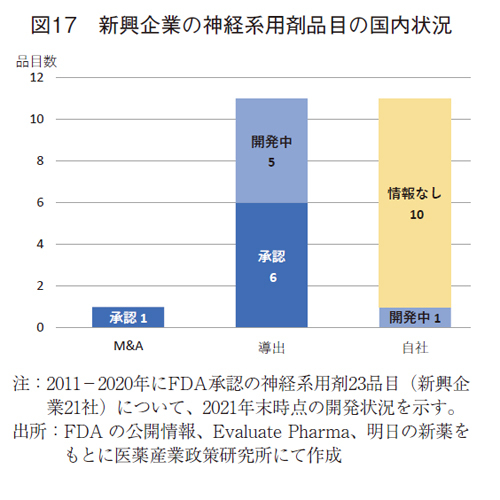
神経系用剤23品目(21社)を分類すると(図17)、抗悪性腫瘍剤とは異なり、M&Aは1社のみであった。導出は11品目でみられ、いずれも承認あるいは開発されていた。M&Aや導出以外は自社の分類となるが、11品目中1品目のみで国内開発が確認された。開発が確認されなかった残りの10品目では国内で臨床開発が実施されている情報は確認されなかった。
新興企業による神経系用剤の開発経緯は、抗悪性腫瘍剤と異なっているようであった。すなわち、新興企業が新薬を創製し、研究開発を進めた品目というより、他企業から早期段階で新薬候補品を導入し、米国を中心に臨床開発を進めた品目が多いようであった。情報なしの10品目については、5品目はFDA承認前に他企業から導入した開発品であった。導出入時期や個々の権利を把握するには限界があり、詳細な調査に及んでいないが、米国でのFDA承認を得た新興企業が日本テリトリーの権利を持たない事例も2社見られており、この権利関係が日本での開発が進まないことに影響する可能性も考えられた。
4. 考察
2016-2020年における未承認薬増加の要因を探ったところ、最も影響の大きい因子として、ピボタル試験への日本組入れがないことが挙げられた。特に新興企業のFDA承認品目増加に伴い、新興企業品目のピボタル試験への組入れ率が低いことは大きな要因であった。さらに抗悪性腫瘍剤においては、ピボタル試験の早期臨床相化が見られ、より一層、日本地域の組入れのハードルが上がっていた。一方、製薬企業のピボタル試験では、国際共同治験への日本地域の組入れが各製薬企業により推進され、組入れ率は43%、Phase3では56%に達し、取組みの成果がみてとれた。
新興企業のピボタル試験にはなぜ、日本は組入れられないのであろうか?
新興企業の投資意思決定として、研究開発と期待事業価値の二側面から考察を加えたい。
研究開発:薬事・臨床試験環境
新興企業のほとんどの抗悪性腫瘍剤の品目で国際共同治験を実施していることから一定規模の開発費用は確保されていることは示唆される。その組入れ地域の選択優先度にて日本は低いことが考えられる。韓国、台湾、香港、シンガポールが多く組入れられていたことから、いわゆる地理的なFar Eastの理由ではないであろう。この中でも日本の優先度が低い理由までの調査は及んでいないが、患者登録数・登録速度などの試験期間への影響、試験開始にあたっての手続きの煩雑さ(日本語書類の整備を含め)、臨床試験の費用などが日本地域を選びにくい可能性として考えられる。また、国際共同治験に日本が組み入れられる際には、日本人での忍容性評価等の追加的な臨床試験を事前に行う必要がある。すなわち、追加の費用や時間がかかることを示しており、この影響は甚大である可能性が高い。Phase2が国際共同治験の際には、推奨用量がまだ決まっていないPhase1実施中に、日本においてPhase1を並行実施する必要が想定され、実施規模は膨らむと共に実施難易度は高いことは容易に想像できる。新興企業からみた日本地域における薬事・臨床試験に関わる課題については、より深い精査が必要とされる。
日本展開の期待事業価値
新興企業が国際共同治験に日本を組み入れるためのモチベーションは、日本市場に早く事業展開する場合が考えられる。また、自社ではなく、M&Aや導出による日本開発がなされている現状をみると承認時点の製品の現在価値を高めたい場合であろう。新興企業は米国の企業がほとんどであり、米国内での収益を期待していることが想定されるが、日本等への事業地域の拡大による製品の現在価値を向上させる意向は自然な考えである。しかしながら、ここ10年の日本の相対的な市場規模は縮小し、マイナス成長の市場と映っていることが示唆された。この状況下では日本地域を組入れ、日本テリトリーへの拡大により現在価値を上げるモチベーションは持ちにくいことは想像に難くなく、初期事業展開における日本優先度は低いものと推察される。
新興企業がピボタル試験に日本地域を組み入れていない理由については、公開情報からの推察には限界があり、個社毎の実態聞き取り調査等の必要性が考えられる。
新薬一品目で、新興一企業が日本国内の開発・薬事規制対応・販売・流通等のインフラを構築する投資をするとは経済合理性から考えにくい。現に抗悪性腫瘍剤の三分の二、神経系用剤の二分の一はグローバル企業によるM&Aや日本テリトリーの導出が行われている。すなわち、日本での新薬アクセス確保には、M&Aをするグローバルメガの製薬企業や日本テリトリーに導入する日本国内の製薬企業の日本事業へのモチベーションに期待するところも大きいのであろう。
新興企業品目は、FDA承認の1-2年後にM&Aや導出により日本での事業展開が図られていたケースもかなりあり、その場合には日本でドラッグロスとなることは避けられている。承認後からの事業取引のため、3年以上の開発ラグは発生している現状であり、ピボタル試験前のより早い開発段階から提携を進めることが期待される。全開発品の中で新興企業の品目が8割に上るといわれている中で、成功の可能性の高い品目・新興企業を早期に買収・日本テリトリー導入することは、グローバル製薬企業や国内企業にとって、難易度は高いと思われるが、リスクをとって早期に提携する製薬企業の取組み継続を期待したい。
早期提携するグローバル製薬企業や日本企業においても、日本の医薬品市場の事業的な魅力度は重要であり、日本に最新医薬品のアクセスを確保する上では、経済合理性の側面から魅力の高い日本市場となることは必須であることには違いない。
2010年以前に取り上げられたドラッグ・ラグの課題は、国際共同治験への日本地域参加が製薬企業により推進され、一定の成果を上げたものと考えられる。薬事承認の審査期間についても短縮が図られ、大きく進展した。今回、新興企業の品目が増えたことで、新たなドラッグ・ラグの兆候が示され、その課題はより複雑であることが示唆された。未承認薬増加の主要因である“新興企業のピボタル試験に日本地域が組み入れられない”ことの理由までは明らかにはなっていないが、未承認薬増加を解消するためには、“新興企業品目の国際共同治験に日本地域が組入れられるようにするためにどうするか”あるいは、“日本市場展開を意図する日本・欧米の製薬企業が早くから提携できるか”が命題であることが考えられた。企業努力を超えて、海外の新興企業が日本を組み入れるインセンティブを海外の新興企業に提供する政策や製薬企業が日本で事業展開する際にも収益を担保できるための政策が重要であろう。日本への最新医薬品のアクセスを確保するためには、世界の医薬品開発状況の変化を鑑みた政策の議論が進むことを期待したい。
一方、日本への最新医薬品アクセスを最重要視する際には、日本人データなしで海外での有用性検証のデータでの承認を許容するなどの規制の見直しも必要であるかもしれない。個別化医療が進み、個々人に合わせた薬剤が増える中、民族的要因や環境要因に過剰に留意することなく、有用性が評価できるような科学の進展とともに薬事規制の在り方についても議論が進むことを切に願う。
謝辞
未承認薬増加への影響度を統計学的に評価するにあたり、学習院大学 経済学部 西村 淳一 教授に懇切丁寧にご指導・ご支援頂いた。この場を借りて深く御礼申し上げます。
付表1
-未承認薬増加への影響度-統計解析 方法と結果詳細-
104品目のデータを用いて品目レベルのロジスティック回帰分析を行った。被説明変数は未承認薬であれば1をとり、承認薬であれば0をとるダミー変数とした。説明変数として、当該品目が新興企業品目であれば1をとるダミー変数、ピボタル試験相が早期化されている(フェーズ3より前のフェーズ)のであれば1をとるダミー変数、そしてピボタル試験相への日本組入れがされていないのであれば1をとるダミー変数を用いた。これらの要素以外に、2010年代前後の臨床開発における環境変化等のマクロの影響を考慮するため、時期ダミー(2010年代後期であれば1をとるダミー変数)を推定モデルに入れた。
分析の頑健性をチェックするため、最小二乗法を用いた線形確率モデルによる推定も行った。
-
1)医薬産業政策研究所「ドラッグ・ラグ:国内未承認薬の状況とその特徴」政策研ニュースNo.63(2021年7月)
-
2)健康・医療戦略推進本部「第1回 医薬品開発協議会 資料2-6」(2020年10月27日)
-
3)医薬産業政策研究所「ドラッグ・ラグ:未承認薬は日本のアンメット・メディカル・ニーズに応えうるか?」政策研ニュースNo.66(2022年7月)
-
4)IQVIA“ Global Trends in R&D: Overview through 2020”(May 2021)
-
5)医薬産業政策研究所「抗悪性腫瘍剤と神経系用剤におけるFDA承認動向の変化 -日本の未承認薬増加の背景- 」政策研ニュースNo.66(2022年7月)
-
6)
-
7)医薬産業政策研究所.「近年の国際共同治験の参加国の分析-臨床試験登録システムClinicalTrials.govを基に-」政策研ニュース No.58(2019年11月)
-
8)医薬産業政策研究所「近年における国際共同治験の動向調査」政策研ニュースNo.66(2022年7月)