Points of View 実用化の進む遺伝子治療の現状と将来展望
医薬産業政策研究所 主任研究員 高橋洋介
はじめに
近年、創薬モダリティ(以降、モダリティ)は多様化しており、低分子医薬だけでなく、組換えタンパクや抗体医薬をはじめ、核酸医薬、遺伝子治療、細胞治療など、様々なモダリティが実際に医薬品として実用化されはじめている1)。本稿においては、その中から遺伝子治療に焦点を当て、上市品や開発パイプライン動向、技術開発のトレンド、研究開発上の各種課題(安全性面、製造・品質管理面、規制面など)について俯瞰する。特に本領域への取り組み状況は各国毎に特色があり、それらを比較・検討することで、日本における遺伝子治療の将来展望を筆者なりに考察してみたい。
遺伝子治療(Gene Therapy)という用語は、学術論文や書籍を含め、各種情報メディアにおいて、様々な定義で用いられているようである。広義には「遺伝子を用いた疾患の治療や予防方法」として、ウイルスベクターやプラスミドベクター、mRNAなどによって遺伝子を導入する手法もしくはゲノム編集を達成する手法など、すべてを指して用いられ、生体内に直接投与するもの(in vivo)とこの手法を用いた細胞を生体内に投与するもの(ex vivo)が存在している。一方で、単一遺伝子変異で発症する希少疾患の治療薬などにおいて、元来ヒトが保有する遺伝子を補充する場合に限って遺伝子治療と定義されている場合もあり、外来遺伝子を導入する場合(DNAワクチン、mRNAワクチンなどが該当)には遺伝子治療と定義しない場合もある。実際にFDAでは、モダリティとしては類似のウイルスベクターを用いて遺伝子導入する医薬品であっても、遺伝子治療(Cellular& Gene Therapy Products)とワクチン(Vaccines)は別カテゴリーとして分類されている2)。本稿においてはモダリティ的に類似であることに主眼を置き、広義の遺伝子治療を「遺伝子治療」と定義/表記し、分析・考察を行うこととする。また、遺伝子治療の要素技術を細分化して分析する際には「in vivo遺伝子治療」、「ex vivo遺伝子治療(遺伝子細胞治療)」、「腫瘍溶解ウイルス」の3つのカテゴリーに分類している。
遺伝子治療の研究開発の状況
国立医薬品食品衛生研究所・遺伝子医薬部のホームページによると、2022年1月時点において日本ではin vivo遺伝子治療製品が3品目、ex vivo遺伝子治療製品(遺伝子細胞治療製品)が4品目承認(条件付き承認含む)されている3)。また、広義の遺伝子治療に含まれる、生体内で抗原タンパク質を発現させるワクチンについては、2022年1月時点でCOVID-19に対するワクチンが3品目承認されている4)。これらはいずれも2019年以降に承認されたものであり、今後ますます遺伝子治療製品の実用化例は増加していくと考えられる。
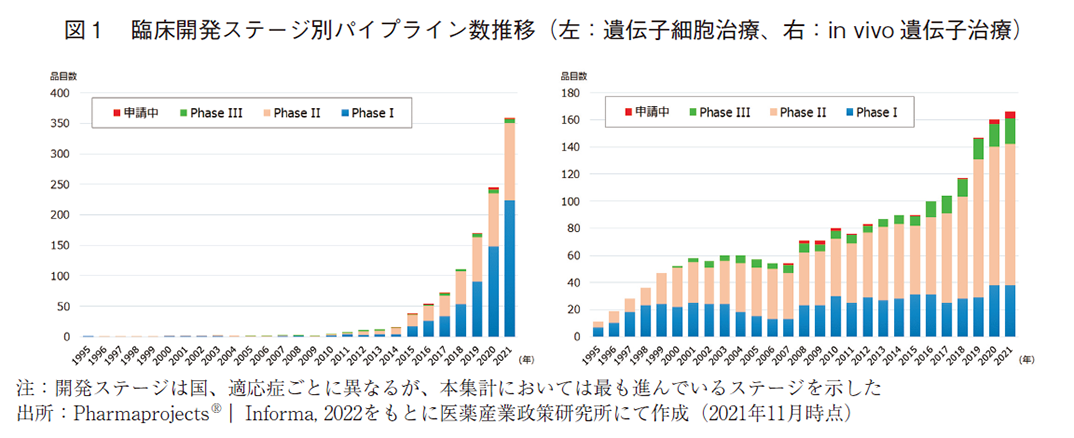
医薬品データベースであるPharmaprojectsを用いて、遺伝子細胞治療およびin vivo遺伝子治療それぞれについて開発パイプライン数の推移を分析し、図1に示した。遺伝子細胞治療は2010年代以降に指数関数的にパイプライン数が増え、2017年にアメリカで2品目のCAR-T細胞が承認されるなど、複数の成功例が出て来ている。この成功を追い風にその後もパイプライン数は伸び続けており、確かな抗腫瘍効果を期待できるモダリティとして、地位を確立しつつある。in vivo遺伝子治療については、1990年代から比較的多くのパイプラインが存在していたが、2000年代にはしばらくパイプライン数が停滞する時期が続いていた。2010年前後になって、パーキンソン病、AADC欠損症、レーバー先天性黒内障、血友病、脊髄性筋萎縮症などに対して臨床的有効性が次々と示されたこと6)がきっかけとなり、それ以降パイプライン数は右肩上がりに増加したと推察される。
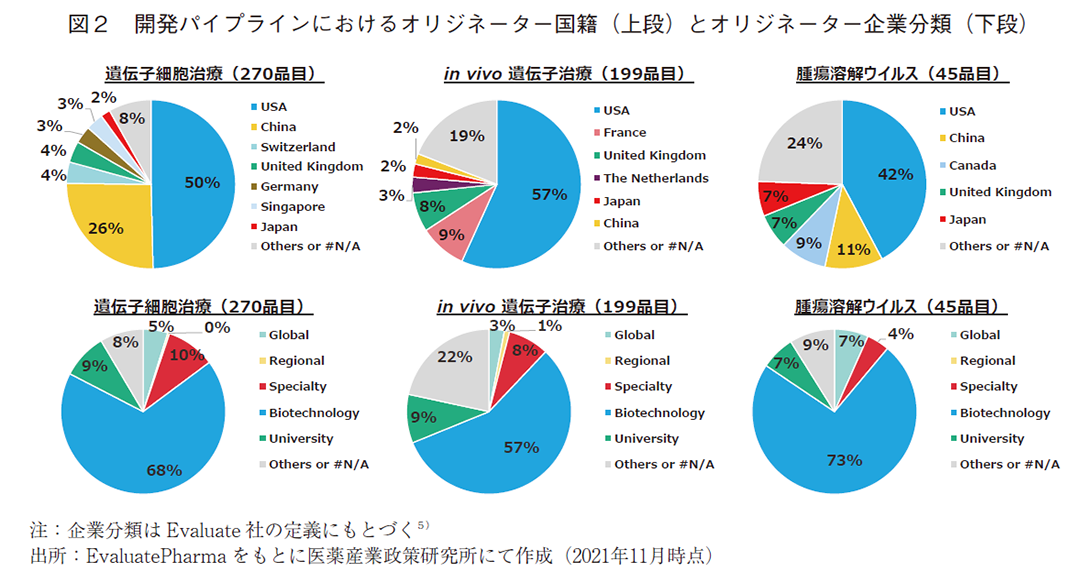
次にEvaluatePharmaを用いて、遺伝子細胞治療、in vivo遺伝子治療、腫瘍溶解ウイルスについて、オリジネーター7)国籍およびそのオリジネーターの種別を分析し、図2に示した。なお、Pharmaprojectsを用いて調査した図1の結果と品目数の不一致が見られるが、これは両データベースの分類の基準やデータ収載対象範囲などが異なるためである。遺伝子細胞治療では、パイプラインの約50%が米国発であり、中国発のものが約26%と、この2国で全体の約3/4を占めているのが特徴的である。中国では様々なCAR-T細胞やCAR-NK細胞など、数多くの抗腫瘍薬がパイプライン上に存在している。2015年に中国が公布した「中国製造2025」において、バイオ医薬は10の重点分野の一つとして位置づけられており、本モダリティを活用して様々な抗腫瘍薬を開発しようと国を挙げて取り組んでいることが窺える8)。in vivo遺伝子治療では、パイプラインの約57%が米国発で、それに次ぐのはフランスやイギリス、オランダなどの欧州各国であり、これらの国ではin vivo遺伝子治療に注力していると推測できる。腫瘍溶解ウイルスでは、全体の品目数はまだ多くはなく、パイプラインの約42%が米国発であり、それに次ぐのは中国の11%、カナダの9%であった。中国は、遺伝子細胞治療と同様に、抗腫瘍効果が期待できる本モダリティに注力していると推測できる。日本に目を向けると、腫瘍溶解ウイルスでは約7%を占めているものの、遺伝子細胞治療およびin vivo遺伝子治療ではともに約2%に留まっており、これらモダリティにおいては海外に遅れをとっていると言わざるをえない。
これらモダリティのオリジネーター種別はいずれも同様の傾向にあり、GlobalやRegionalに分類されるような大手製薬企業由来のパイプラインは少なく、Biotechnology由来が過半数を占めていた。また他モダリティと比べると、Universityの割合が比較的高いのも特徴である。特にin vivo遺伝子治療ではOthers or #N/Aの割合が比較的高くなっており、これはHospitalやNon profitなどに由来するパイプラインが多いことに起因している。またin vivo遺伝子治療では患者数の少ない希少疾患を対象とした開発パイプラインが多く(詳細データは割愛)、これら疾患の治療薬創製においては、製薬企業以外の主体が重要な貢献を果たしている現状が見て取れた。
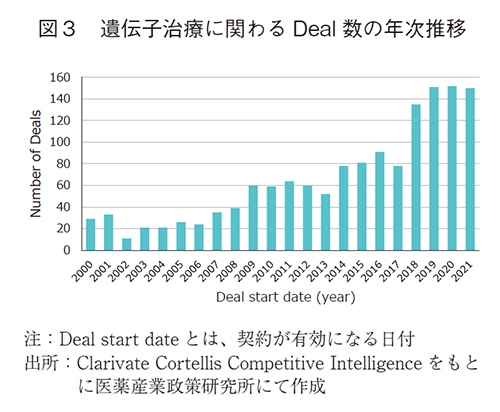
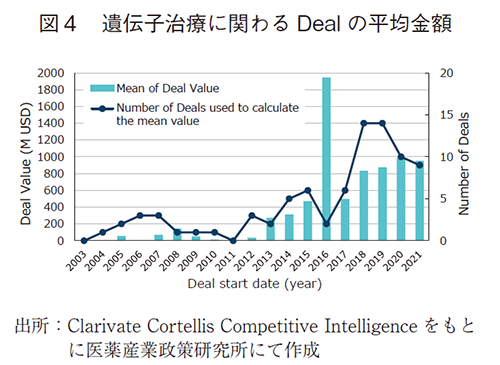
スタートアップ企業やアカデミアから創出された遺伝子治療のシーズは、規模の大きな製薬企業への導出もしくは共同開発によって実用化を目指すケースが多い。この状況を把握するため、Clarivate Cortellis Competitive Intelligenceのデータベースをもとに、遺伝子治療に関わるDeal数(図3)やDeal金額(図4)、Dealの国際間動向(図5)について調査を行った9)。Deal金額の調査については、Productの権利移管に関わる取引に限定し10)、Total Projected Current(契約の総額(現在予想額))について各年毎の平均値を算出した。図3に示した通り、遺伝子治療に関わるDeals数は年々増加しており、本モダリティへの注目度が年々高まっていることが窺える。図4に示したDeal額については、金額が公表されているものに限った平均値であることに留意する必要はあるが、年々高騰しており近年では平均値として10億ドルに迫るほどである。DiscoveryやPreclinicalの段階のProductが多い中(個別データは割愛)での平均取引額の高さは、対象のProduct単体に対する評価だけでなく、Productに関わる基盤技術に対して高い評価がなされているからと推察でき、有望なシーズや基盤技術に対する投資が過熱していることが見て取れる。
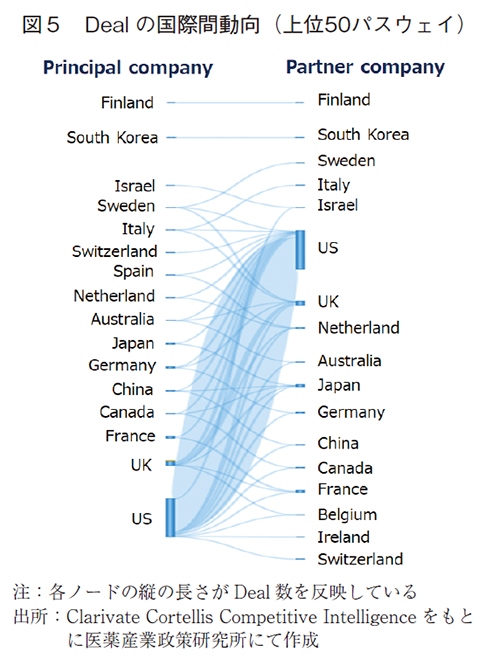
図5に示したDealの国際間動向(上位50パスウェイ11))では、Principal company(シーズ創出側)の国籍、Partner company(シーズ取り入れ側)の国籍ともに米国が大半を占めていた。Principal companyとしてもPartner companyとしても米国につぐのは英国であり、特にPartner companyとして様々な国からシーズを積極的に取り入れていることが特徴的である。日本に関しては、日本国内でのDealや、米国とのDealが中心であった。
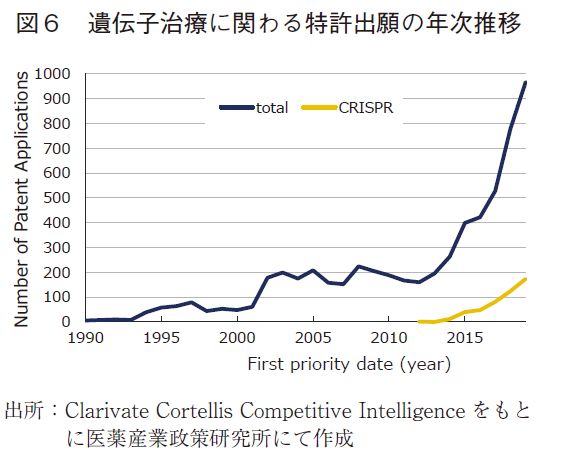
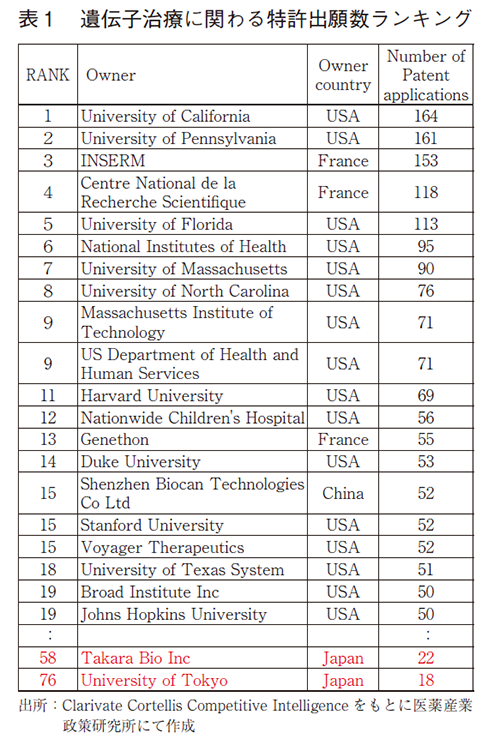
次いで、パイプライン創出の源泉となる特許出願の動向について調査した。Clarivate Cortellis Competitive Intelligenceのデータベースをもとに、遺伝子治療に関わるPCT出願を抽出するとともに、近年注目されているゲノム編集技術のCRISPR-Casに関わるPCT出願動向を調査した。具体的には、検索式Any Action(Gene therapy)AND Technologies(Gene transfer system)によって遺伝子治療に関わる特許出願を抽出し、このうちPCT出願されたものを分析対象とした。また特許数の年別推移(First priority date、優先日にて分類)に関しては、検索結果のTarget-based Actionsの項に「CRISPR」の文字を含むものも抽出し、CRISPR-Casシステムを用いたゲノム編集に関わる特許出願数として、あわせて結果を図6に示した。また、特許出願人(Owner Company)に関して、出願数に基づくランキングを作成し表1に示した。なお、一つの特許出願に関して複数の出願人が存在する場合には、それぞれを1件としてカウントした。
図6の通り、遺伝子治療に関わる特許出願は、1990年頃にはほとんど存在していなかったが、2000年前後にかけて大きく増加し、200件/年ほどに到達していた。しばらくはそこから一定数を維持していたが、2010年代になり遺伝子治療の臨床試験で大きな成果が報告され始めたことなどを背景に、近年ではさらに急速に増加している。また2012年に開発されたCRISPR-Casシステムに関わる特許出願は、それ以降著しい伸びを見せており、この技術を応用した研究開発が盛んになっていることが特許出願数からも推測できる。表1の出願人ランキングを見ると、上位に位置するのはほとんどが米国の企業や研究機関であり、ついでベスト20に3者がランクインしたフランスが存在感を放っている。また中国からも1者がベスト20にランクインしている。一方で、この切り口で分析した場合には日本の存在感は相対的に薄く、ベスト100まで見ても2者がランクインしたのみであった。
日本における遺伝子治療の課題
ここまで見てきたように、近年急速に実用化の進む遺伝子治療であるが、日本での研究開発が海外に対して遅れをとっている事実に対して、様々な課題があると指摘されている。健康・医療戦略推進本部の第5回ゲノム医療実現推進に関するアドバイザリーボードでは、各構成員から様々な課題や対応策が提示され議論が交わされている12)。そこでの議論内容を参考に、筆者なりに各種課題を分類・整理し、表2に示した。
表2の通り、遺伝子治療の研究開発を推進する上で、様々な課題が山積している。基礎研究の充実という観点では、有効性や安全性を一層向上し遺伝子治療の産業化を促進するため、モダリティをさらに洗練していく必要がある。これは日本特有の課題ではなく世界共通の課題ではあるが、日本では遺伝子治療に対する研究者数が相対的に少なく、結果として基礎研究で海外に後塵を拝している点において、日本での課題感は大きい。この状況を打開するため、遺伝子治療に関わる研究の重要性を広く世に周知し、政府からの支援やファンド等からの投資を活性化して長期的に研究費を充実させ、基礎研究に従事する研究者数を拡充することが重要となるだろう。こうして研究が進展し、やがて日本初の実用化例が出始めると、健康・医療の分野での産業化の道筋が確立され、さらに投資を呼び込む引き金となり、結果的に研究が一層活性化するという、好循環へとつながっていくのではないだろうか。

また、遺伝子治療においては、バイオ医薬品分野に特有の特許戦略の重要性が指摘されている。物質特許によって医薬品の権利を包括的に主張出来る低分子医薬品とは異なり、バイオ医薬品では複数の様々な特許の組み合わせによって医薬品の権利を確保することが重要であり、自らの特許出願を含む、クロスライセンスなどの戦略的な特許戦略によって国際競争力を保持するため、リソース(特に人材、資金)の確保・拡充が必要である。この点について、シーズ創出元として期待されるアカデミアやスタートアップなどが個々に対応していくことは困難なケースが多く、国立研究開発法人日本医療研究開発機構(AMED)等の公的機関や連携先の企業などが早い段階から支援していくこと、場合によってはプレイヤー間で特許を共有するような仕組み作りなどが重要となるだろう。
ここまで課題が山積していると述べてきたが、実際には積み残しされているわけではなく、これら課題への対応策は講じられつつある。さらにコロナ禍を経て、このモダリティが感染症領域のワクチンに応用可能であったことも追い風となり、加速度的に課題対策が進み始めている。以降では、「規制の整備・緩和(特にカルタヘナ法関連)」、「製造施設の拡充」、「基礎研究の充実(特に安全性面上の課題)」について、具体的な動向を示していきたい。
カルタヘナ法を巡る規制の動向
日本で遺伝子治療の開発を進める場合、多くのケースで「遺伝子組換え生物等の使用等の規制による生物の多様性の確保に関する法律(カルタヘナ法)」に関わる対応が必要となる。
カルタヘナ法は、2000年1月に採択された「生物の多様性に関する条約のバイオセーフティに関するカルタヘナ議定書(カルタヘナ議定書)」という国際ルールに基づいて、日本国内で制定された法律である。日本国内で遺伝子治療の研究開発を進める上で必要となる、カルタヘナ法に係る対応について要約すると、以下の通りである。閉鎖系において拡散防止措置下で遺伝子組換え生物等を用いる場合(大腸菌を用いてプラスミドベクターを製造する場合など)には「第二種使用等」に該当し大臣確認(もしくは機関内承認)が必要となり、遺伝子組換え生物等(組換えウイルスなど)をヒトに投与する等、開放系で用いる場合には「第一種使用等」に該当し大臣承認が必要となる。これら詳細については、PMDAウェブサイト上の「カルタヘナ法に係る申請」の説明ページで分かりやすくまとめられているため、そちらを参照いただきたい13)。また日本製薬工業協会ホームページ上では、バイオ医薬品委員会の成果物として「遺伝子治療用製品等及び感染症の予防を目的とする遺伝子組換え生ワクチンの治験実施までの留意事項」が公開されている14)。遺伝子治療の研究開発に必要な知識や、効率的に開発を進めるための情報など、非常に分かりやすく整理されている。
カルタヘナ議定書は、生物の多様性の保全を目的として定められた国際ルールであり、考え方としては大変意義深いものと考えられるが、一方でアメリカなど一部の国ではカルタヘナ議定書を批准していないなど国際的な足並みが揃っていない。この原因は、遺伝子組換え生物等によってもたらされるメリットと、生態系への悪影響リスクに対して、バランスの取れた着地点を模索し国際間で合意を得ることが困難であったことにある。このような経緯はあるが、結果的に日本で遺伝子治療の開発を進める上では、アメリカでは不要である上記のような対応が必須となり、これが日本での開発遅延や研究開発の阻害要因につながってしまっているという側面がある。
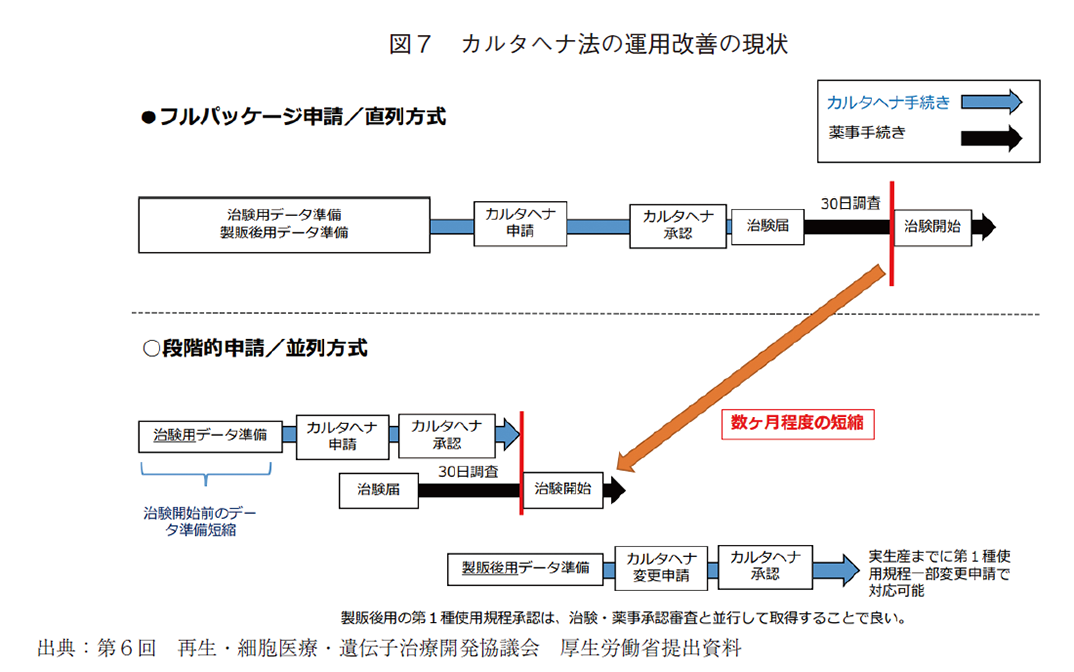
上記のような背景から、カルタヘナ法による規制が日本での遺伝子治療用製品等の研究開発の足枷となっているという意見も散見されるが、本課題については産学官の共通認識のもとに議論が進められて来ており、課題解決へ向けて着実に前進している。従来、治験開始前の段階から承認後の実生産規模を見越したフルパッケージの申請が必要であった点については、事実上の一部変更手続きを明確化した。また、本邦と同様にカルタヘナ議定書に批准している欧州での事例を調査し、治験開始前には必要最小限のデータで承認を受けたのち、治験を実施しながら必要なデータを段階的に準備することが可能となるよう、産学官での検討が進められている15)。治験届を提出する前に第1種使用規程承認を受ける必要があった点は、「治験を開始する日まで(具体的には、「当該医薬品等の治験に係る最初の国内被験者組入れ(治験参加登録)まで)に承認を受ければよいことになった16)。これらについては、図7に示した通り、第6回再生・細胞医療・遺伝子治療開発協議会における厚生労働省提出資料において分かりやすくまとめられているのでそちらを参照いただきたい17)。
また、研究目的で製造された第二種使用等確認した製品(文部科学大臣確認済)に関して、製造場所・製造方法・製造量等に変更ない限りは、産業目的で製造する場合に改めて厚生労働大臣による確認を受ける必要がないことが明確化されている18)。
このように、これまでに蓄積されてきた知見や海外での規制状況に基づき、カルタヘナ法の基本的な考え方を尊重した上で様々な運用改善が行われ、研究開発に要する期間は短縮されつつある。今後のさらなる議論を経て、日本での研究開発を推進する上でのより理想的な着地点が見出されることに期待したい。
製造施設の拡充における課題
遺伝子治療製品の製造施設や製造に関わる専門人材の不足という課題に対して、国内における取組に関していくつか紹介する。
次世代バイオ医薬品製造技術研究組合(MAB組合)は、2013年に設立された技術組合であり、バイオ医薬品の製造技術開発を目的として大学や企業の研究者などが参画している。従来は抗体医薬の製造技術開発に注力していたが、2018年度からはAMEDの「再生医療・遺伝子治療の産業化に向けた基盤技術開発事業(遺伝子治療製造技術開発)」に採択され、研究開発課題「遺伝子・細胞治療用ベクター新規大量製造技術開発」がスタートしている。本研究成果として、現在ボトルネックとなっている遺伝子治療用ベクターの大量製造技術/分析技術や、治験実施に向けた規制対応方法などが、将来的には日本の強みとして確立されることを期待したい。
また、製薬協の政策提言をきっかけとして、神戸大学及びMAB組合所有のGMP準拠製造施設を活用し、2017年にバイオロジクス研究・トレーニングセンター(BCRET)が設立されている。BCRETでは、製薬企業等に対して遺伝子治療製品の製造開発に関わる教育トレーニングや、審査やGMP適合性調査などに関わる審査官等に向けた研修を実施しており、特に人材育成の観点から遺伝子治療製品の研究開発の推進の役割を担っている。
政府の動きで予算規模の大きなものとして「ワクチン生産体制強化のためのバイオ医薬品製造拠点等整備事業」があり、2021年度補正予算案で2274億円が計上されている。この事業はその名の通り、有事(感染症流行時等)にワクチン生産を速やかに行うことが出来る製造施設の整備を目的としたものであるが、平時にはバイオ医薬品の製造を行うことが可能なデュアルユース設備とすることが掲げられている。この製造施設が稼働する際には、新型コロナウイルス感染症が収束し、遺伝子治療製品をはじめとしたバイオ医薬品の製造に最大限活用されることを願いたい。
では、海外ではどのように遺伝子治療分野の製造に関わる課題に対応し、研究開発が推進されているのであろうか?参考になる事例として英国と米国の事例を取り上げたい。
英国では、2010年に「カタパルト構想」が提唱され、将来の英国の経済成長を牽引するであろう重要分野に対して、それぞれの分野に対して産学連携拠点(カタパルト)を整備し、集中的に予算を投入する国家主導のプロジェクトが推進されている。このプロジェクトの1つとして2012年に設立されていたCell Therapy Catapultを発展させる形で、2018年にCell and Gene Therapy Catapult(CGT Catapult)が設立されており、本領域の研究開発において中心的役割を担っている19)。
CGT Catapultのビジョン・使命として、(1)産学ネットワーク構築によるイノベーションの実現、(2)イノベーションの商業化、(3)独自の研究・製造施設や専門知識で産学連携を補完、(4)産業界、研究機関、政府、保健医療機関、業界団体、国際機関等との連携による英国のエコシステムの成長を促進、以上の4点が掲げられている。このプロジェクトで特に特徴的なのは、拠点であるStevenageに大規模なGMP製造施設を建設している点である。この製造施設は英国政府による約6,000万ポンドの投資により建設された後、それ以後も断続的に施設の拡張が行われている19)、20)。新規技術である遺伝子治療では、研究開発における不確実性が特に高いだけでなく、経験を積んだ専門人材の少なさや研究開発初期から高額な製造コストを要することが高い参入障壁となり、産業としての実用化の阻害要因となる。CGT Catapultではこの点が強く意識されており、利用者のニーズに応じて、治験薬の製造をはじめとして、製造施設を柔軟に共同利用できる仕組みを構築するとともに、人材育成の場と捉え多くの人材を雇用・育成することで、細胞治療・遺伝子治療を将来の英国の主要産業へと発展させようという意図が見て取れる。CGT CatapultのAnnual Review 2021によると、直近では3,800人の専門人材のスキルアップに貢献するとともに、132のプロジェクトが進行しているようである(図8)21)。

米国での取り組みで注目すべきものとしてPaVe-GT(Platform Vector Gene Therapy)22)とBGTC(Bespoke Gene Therapy Consortium)23)を取り上げたい。
PaVe-GTはNIH内に設置されているNCATSが主導するプロジェクトであり、4つの異なる希少疾患(有機酸血症2種(PCCA deficiency、MMAB deficiency)、先天性筋無力症候群2種(DOK7 deficiency、COLQ deficiency))に対する遺伝子治療の研究開発を行うものである。本プロジェクトにおいては、共通のカプシド(AAV9)や共通の製造方法・精製方法によって候補製剤を創製し、共通の非臨床試験やCMC評価手法の採用、マスタープロトコールにより臨床試験を効率的に進めるなど、標準化された手法を用いることで遺伝子治療の研究開発の効率化の可能性を検証することを目的としている。さらには、一連の研究成果や米国食品医薬品局(FDA)とのやり取りなど、様々な情報を広く公開することで、将来的なAAV遺伝子治療の技術開発に有用な知見をもたらすことを意図している。
BGTCは、前述のNCATSなど複数のNIHの研究機関、FDA、製薬企業10社、患者団体など、計27の様々なステークホルダーが参画するコンソーシアムであり、2021年10月に発足のプレスリリースがなされた24)。5年間で7,650万ドルの予算規模であり、AAVを用いた4~6件の臨床試験の実施を通して、合理的かつ効果的な標準的評価項目や評価方法を確立するとともに、規制のあり方、均一な製造プロセスなどについても検討される。そして希少疾患のための遺伝子治療の研究開発コストの低減可能性を模索し、商業的実現性や持続性への道筋を作ることも目標として掲げられている。
これら海外の事例からの学びとしては、政府のリーダーシップのもとで積極的な投資がなされ、研究開発推進上のボトルネックとなる部分を解決するべく、多くのプレイヤーが参画し有機的に連携しながらプロジェクトを進めているという点である。単に空間的に近い距離に集まるだけではなく、一つのプロジェクトに、共通のビジョンのもとに多くのプレイヤーが集い、心理的に近い距離間で協働する、そのようなエコシステムの形成が特徴であり重要なポイントと言えるのではないだろうか。
安全性上の課題
遺伝子治療の研究開発の歴史上、安全性面での課題が常に付きまとってきた。1999年に開始されたX連鎖重症複合免疫不全症(X-SCID)に対する造血幹細胞遺伝子治療において、世界で初めて遺伝子治療単独で明瞭な薬効が認められたものの、その後治療を受けた患者で次々と白血病を発症することが報告され、遺伝子治療全般の研究開発に深刻な影響を与えた。その後の研究において毒性メカニズムが明らかとなり、この時用いられたレトロウイルスベクターにより、意図せぬ部位に遺伝子挿入されることで発がん性リスクが上昇することが明らかとなった。近年では改良型レトロウイルスベクターも開発され、本リスクは著しく低下したとの報告もある6)。
また、近年ではアデノ随伴ウイルス(AAV)ベクターを用いたin vivo遺伝子治療の研究開発が盛んに行われている。AAVベクターは、非病原性ウイルス由来であり安全性が高いこと、非分裂細胞に対しても比較的高い遺伝子導入効率を示すこと、組織指向性の異なる血清型が複数存在し生体内の様々な臓器をターゲットに出来ることなどから、in vivo遺伝子治療用ベクターとして汎用されている。ただし、AAVにおいても臨床試験でいくつかの毒性が認められており、安全性面での課題は依然として残されている。安全性面における課題は遺伝子治療分野では世界共通の課題であるが、米国では世界に先駆けて課題解決に向けた議論や取り組みが行われている。2021年9月には、FDAにおいてCellular, Tissue, and Gene Therapies Advisory Committee(CTGTAC)Meetingが開催され、AAVの安全性リスクに関して集中的な議論が行われた25)。臨床試験で認められている重篤な副作用としてOncogenicity Riskの他、表3に示したHepatotoxicity、TMA(Thrombotic Microangiopathy)、Neurotoxicityが取り上げられ議論されている26)、27)。これら毒性のメカニズムについては色々と仮説は提唱されているものの、まだ明確な結論は出ていない。遺伝子治療では、安全性評価における動物モデルの限界や、品質面の担保(空のカプシド等を含む、種々の不純物による毒性評価)などで課題が残されているが、今後のさらなる基礎研究・応用研究の進展により、これら安全性面の課題が克服され、遺伝子治療の研究開発が一層加速することを期待したい。

おわりに
ここまで、遺伝子治療に関わる基礎研究から実用化研究・臨床試験の動向を概観し、海外と対比しながら日本の現状と課題そして課題対応策について考察した。海外に対して遅れをとっている現状が明らかになる一方で、この遅れを挽回するべく日本国内で様々な策が講じられつつある姿も見えてきた。基礎研究の進展や充実の必要性という点では、これらは依然として世界共通の課題であり、医療応用のために遺伝子治療を一層確度の高いモダリティへと洗練していくため、さらなる科学的・技術的なブレークスルーが求められる。具体的には、遺伝子発現効率の向上(投与量の低減)、遺伝子発現の制御(発現量や発現時間の最適化)、毒性メカニズムの解明と毒性回避手法の確立、標的指向性の付与(全身作用型や組織選択型のデリバリー)、細胞培養方法を含む高品質な製造方法の確立、またこれら課題を総合的に解決することによる治療に必要な医薬品製造コストの大幅な低減など、を解決する必要があると考える。これらのハードルを越えることが出来れば、基礎研究成果が業界内での競争力の源泉になるとともに、場合によっては現状では生涯飲み続けなければならない医薬品を一度きりの投与で治療することが可能な遺伝子治療へと置き換えられるなど、他モダリティに対する優位性獲得にもつながるかもしれない。以上のことから、遺伝子治療の研究開発の推進は、製薬企業の成長や製薬産業全体の発展においてキードライバーとなりえるのではないだろうか。
最後に「なぜ“日本で”遺伝子治療の研究開発を推進するべきか」について、日本の将来のあるべき姿を考察してみたい。1点目は、「日本特有の遺伝子変異に基づく疾患に対する遺伝子治療は、日本自ら開発する必要がある」ということである。海外での研究開発成果に頼っていては、従来型のヘテロな患者層に広く使用可能な医薬品とは異なり、国内の患者層向けの治療薬は創製されないことも想定され、ドラッグラグよりも深刻なドラッグロスへと繋がりかねない。2点目は、「遺伝子治療の技術は、希少疾患だけでなく様々な疾患へと応用可能」ということである。希少疾患への応用だけであれば、多大な失敗リスクを伴う研究開発投資に見合う利益が得られない可能性がある。しかし、希少疾患に対する治療薬の研究開発では、製造面や臨床試験の規模を比較的小さく抑えることが可能であり、この一連の研究開発の経験を糧にして、より大規模な患者層を対象にした治療薬の創製へとスケールアップ/ステップアップしていく流れは、ビジネス的観点でも合理的/効果的ではないだろうか。特に重要な点として、「本技術は新興感染症のワクチン開発にも応用可能」ということが挙げられる。新型コロナウイルス感染症に対する国産ワクチンの開発の遅れを反省材料として、有事の際には速やかに国産ワクチン開発に着手できるように技術開発を進めるべきであり、日本国民の生命や健康を日本の技術を用いて守ることは、国家安全保障の観点からも重要な施策となるだろう。
-
1)医薬産業政策研究所、「新薬における創薬モダリティのトレンド -多様化/高分子化の流れと、進化する低分子医薬-」、政策研ニュース No.64(2021年11月)
-
2)FDA、「Vaccines, Blood & Biologics」(2022/1/12参照)
-
3)国立医薬品食品衛生研究所・遺伝子医薬部、「承認された遺伝子治療製品」(2022/1/28参照)
-
4)PMDA、「PMDAにおける新型コロナウイルス感染症対策に係る活動について」(2022/1/31参照)
-
5)「Global」:グローバルに展開している業界最大手の製薬会社。独自の研究開発や、同業他社の買収や品目のライセンス導入などによって製品ラインナップを幅広く拡充し、グローバルで製品の販売を手掛ける。「Regional」:業界大手の製薬会社。独自の研究開発や、同業他社の買収や品目のライセンス導入などによって製品ラインナップを拡充し、特定の地域において製品の販売を手掛ける。「Specialty」:独自に研究開発を行う製薬会社で、新規有効成分(NME)の研究開発だけでなく、適応拡大や剤型変更など様々な戦略を駆使して製品ラインナップを拡充する製薬会社。単一もしくは限られた治療領域に特化したビジネスを展開する。「Biotechnology」:新規有効成分(NME)の医薬品の研究開発を行っている会社。市販品はなく、研究開発中のプロダクトのみを有する小規模な会社も含まれる。必ずしも(科学的な定義に基づく)バイオテクロノジー製品を開発している企業に限定されない。「University」:一般的に斬新な学術研究を行っている高等教育機関。「Others or #N/A」:Generic、CRO、Government Agency、Hospital、Non-Profit などを含むその他の企業分類、もしくは分類不可の企業。
-
6)小澤敬也/編、実験医学増刊、「いま、本格化する遺伝子治療」、羊土社、2020
-
7)オリジネーターとは、Evaluate Pharma 上に収載されている各プロダクトの開発起源企業
-
8)科学技術振興機構・研究開発戦略センター、「「中国製造2025」の公布に関する国務院の通知の全訳」(2022/1/31参照)
-
9)検索条件:Technologies(Gene transfer system)OR Title/Summary(""gene therapy"")とした
-
10)検索条件:Agreement type(Drug - Development/Commercialization License)とした
-
11)どこの国からどの国へと権利が移っているのかを一つのパスウェイとし、それぞれのパスウェイの数を集計し、上位50パスウェイを図5に示している
-
12)健康・医療戦略推進本部、「第5回ゲノム医療実現推進に関するアドバイザリーボード」(2022/1/12参照)
-
13)PMDA、「カルタヘナ法に係る申請」(2021/12/22参照)
-
14)
-
15)
-
16)
-
17)厚生労働省、「再生・細胞医療・遺伝子治療分野に関連する規制・制度について」(2022/1/12参照)
-
18)
-
19)Cell and Gene Therapy CATAPULT(2022/1/25参照)
-
20)国立研究開発法人 日本医療研究開発機構委託調査、「2019年度 再生医療・遺伝子治療の市場調査業務」(2022/1/28参照)
-
21)
-
22)Platform Vector Gene Therapy website(2022/1/25参照)
-
23)
-
24)
-
25)
-
26)
-
27)